■ Q&Aコーナー > その他全般 |
 |
8.71 無痛化剤などの人体に対する効果が期待される添加剤の投与量はどのように規定されるのか |
|
A: |
一般的に、過去の投与実績から添加剤の投与量は規定される。
ただし、使用前例のない場合、または、使用前例があっても投与経路が異なる若しくは前例を上回る量を使用する場合には、安全性に関する情報を提示する必要がある。
(ICHガイドライン、M4S:CTD-非臨床に関する文書の作成要領に関するガイドライン)
さらに、添加剤の添加量が使用実績以下であっても、添加剤が有効成分の有効性や安全性に影響を及ぼすと推測される場合、事前にPMDAとの相談が必要となる場合もある。 |
回答:--- |
|
|
|
|
8.70 リポソーム製剤などのキャリアを含む製剤では、キャリアの物理化学的な安定性など留意すべき評価項目は何か |
|
A: |
リポソーム製剤に関して、その物理化学的性質(リポソームの形態、電荷、リポソーム中の容積、粒子径、相転移温度、分光学的データ、浸透圧、光散乱指数など)の詳細な評価が示されるべきであるとされている。
粒子サイズなどの物理化学的性質の変化は薬物の放出等に影響を与える可能性があるため、それらの性質に関わる品質の安定性については評価法を確立する必要がある。
製造法等の変更によって粒子径などの物理化学的性質に変化が見られた際には、生物学的同等性試験により品質を担保する場合もある。
参照ガイダンス
FDA : Guidance for Industry Liposome Drug Products (Draft guidance)
EMA : Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product |
回答:--- |
|
|
|
|
8.69 小児用製剤という言葉がありますが、製剤を設計するうえで成人用製剤と比較してどのような要件が求められるのでしょうか? |
|
A: |
小児用製剤では、正確な投与が可能であること、服薬しやすいことが求められます。経口製剤では、種々の年齢に応じて液剤、ドライシロップ、チュアブル錠、口腔内崩壊錠やソフトカプセルなど様々な形態が望まれるほか、主薬の苦味をマスキングする工夫が望まれます。また、国によって異なった種類の製剤、味や色が好まれます。種々の製剤において小児専用の薬物含有量が必要なこともあります。注射用製剤では、投与量が正確かつ安全に投与されるような適切な濃度の製剤や小児の用量に適切な一回投与の包装形態が求められます。坐剤など代わり得る投与ルートやドラッグデリバリーシステムの開発も必要な場合があります。 |
回答:--- |
|
|
|
|
8.68 2013.11.20 薬事法の一部改正が成立したと聞きましたが、ポイントを教えてください |
|
A: |
2013.12.13に公布された「薬事法の一部改正」ポイントはつぎの内容となっています。
- 医薬品販売規制に関する改正
・一般用医薬品は適切なルールの下にネット販売が可能となりました。但し第一類医薬品はこれまでどおり薬剤師が販売します。
・新たに要指導医薬品(スイッチ直後品目・劇薬)を指定し薬剤師が対面販売を行います。
・医療用医薬品はこれまでどおり薬剤師の対面販売
- 指定薬物についてその所持と使用が禁止されます。(学術研究等は除きます)
- この改正は2014.6.12から施行されます。
|
回答:--- |
|
|
|
|
8.67 iPS細胞とES細胞の違いについて教えてください |
|
A: |
iPS細胞もES細胞も多分化能を持つ幹細胞(Stem Cell)です。幹細胞とは様々な細胞に分化する元になる細胞です。この2種類の細胞はその作製方法が異なります。iPS細胞(induced Pluripotent Stem Cell;人工多能性幹細胞)は体細胞に特殊な遺伝子を導入し作製します。ES細胞(Embryonic Stem Cell; 胚性幹細胞)は受精後の胚盤胞から細胞を取り出し培養して作製します。なお「再生医療等の安全性の確保に関する法律」が2013.11.27に公布されました。今後の再生医療などへの応用が期待されています。 |
回答:--- |
|
|
|
|
8.66 製剤製造工程における「Blending」と「Mixing」はどちらも混合と訳されるが、何か違いがあるのか |
|
A: |
英文の専門的な解釈とは異なるかもしれませんが、一般的には、「blend(blending)」という単語は、二種類以上の物質の分量を意図した割合で均質に混ぜ合わせること、一方で「mix(mixing)」は、必ずしも割合や均質性は要件ではなく、単に二種類以上の物質を入れ混ぜる、かき混ぜるといったニュアンスで使い分けがされることが多いのではないかと思われます。
なお、製剤の単位操作としての「混合」工程は、粉末の均一性またはランダム性を得るために、粒子の相対的位置を変更する操作と解説されています(「GMP・ICH医薬用語辞典」(株式会社じほう))。
よって、あくまでも個人的見解となりますが、作業性などの理由から投入の前に予め原料を混ぜておく(Premix)といった場合の混合については「Mixing」、打錠用顆粒などにおいて、主薬や添加剤などの均一性が求められる場合の混合については「Blending」という訳が考えられます。 |
回答:--- |
|
|
|
|
8.65 PIC/SはPharmaceutical Inspection Co-operation Schemeの略称であるが、「Inspection」と「Audit」は、どう違うのか |
|
A: |
辞典によると、「Inspection」は(公式・正式の)視察・監察・検閲・査閲、「Audit」は(会社などの)監査と訳されています(「新英和中辞典」(研究社))。また、「査察」は情況を視察すること、「監査」は監督し検査することと意味付けられています(「広辞苑」(岩波書店))。
Inspectionは主に行政などの公的機関による実地での検査、Auditは主に企業間において(しばしば実地を含む)聴聞による状況確認というのが、一般的な理解ではないかと考えられます。
ただし、PIC/S中の「自己点検(Self Inspection)」や、ICH-Q7A中の「内部監査(Internal Audit)」といった用語や、医薬品の申請時に当局により実地で行われる検査は、適合性査察ではなく「適合性調査」とされることなど、これらの用語の違いは完全に明確にはなっていないようです。 |
回答:--- |
|
|
|
|
8.64 特許の差止請求訴訟等の問題の中で、2013年に米国の特許法が大きく改正されたと聞いたが、どのように変わったのか |
|
A: |
医薬品をはじめ、多くの製品について世界最大の市場である米国における特許制度は、日本を含む他の国の制度と異なる部分があり、その1つに「先発明主義」と「先願主義」があります。先発明主義とは実際の「発明日」を基準に新規性を判断するものであり、一方の先願主義とは、特許の「有効出願日」を基準に新規性を判断するものです。米国は今まで先発明主義を採用していましたが、2013年3月に施行された米国特許法(America Invents Act, AIA)102条の改正により、日本と同様の先願主義に変わることになりました。その他、他者権利への対抗手段や、出願手続きについても変更となっています。
(参考:日本知的財産協会ウェブサイト「米国特許法改正 America Invents Act (AIA)の概要」( http://www.jipa.or.jp/topics/aia_fifth.pdf)) |
回答:--- |
|
|
|
|
8.63 外部委託製造が増加する中、海外で製造し国内に輸入される医薬品も多くあるが、製造される地域や国内外の違いによって、品質上のギャップが生じることはあるか |
|
A: |
近年、医薬品製造技術が専門化する一方でコスト競争はますます厳しくなり、製造コスト削減や設備投資抑制などを目的とした外部委託製造が増加しています。
本学会の会員企業の皆様に対するアンケート調査では、外部委託製造を海外の医薬品製造受託機関(Contract Manufacturing Organization, CMO)で行う場合などにおいて、90%の企業が国内と海外との間で品質ギャップを感じたことがあると回答されており、製造される地域に関わらず、海外よりも国内で求められる品質基準(特に外観について)が一般的に厳しい傾向にあるという意見が多く寄せられました。
アンケート調査結果の詳細につきましては、2013年9月の本学会誌に掲載されておりますので、ご参考下さい。 |
回答:--- |
|
|
|
|
8.62 最近監査などにおいてHSEが話題になるのですが、HSEとは何か、具体的に教えてください。 労働衛生・安全・環境(HSE:Health, Safety and Environment) |
|
A: |
HSEとはHealth(健康)Safety,(安全)Enviroment(環境)の頭文字で、EHSという場合もあります。
広い意味では、装置やシステムが持つ人や社会や環境に対する潜在リスクを分析評価し,それに対応して安全を実現する適切でかつ経済的な手段を採用し、またそこでは使用する設備の何十年にもおよぶ全ライフサイクルを考慮し,採用した手段を正しく機能させる適切な管理を行う、リスクベースドアプローチによるHSEマネジメントという意味でもとらえられます。 |
回答:--- |
|
|
|
|
8.61 ニューラルネットワークがいろいろな論文に出てきますが、なぜこれが製剤開発で、有用なのでしょうか |
|
A: |
人工ニューラルネットワーク(ANN)は、脳の神経細胞網の構造をコンピューター上に模倣した計算システムです。事前に計測された入出力データによってANNを学習させると、未知の入力因子から出力特性を高精度に予測することができるようになります。従来の線形回帰式等に比べて予測精度が高く、多変量データの複雑な因果関係を近似できるという大きなメリットがあります。このため、製剤設計で計測した多変量データを用いてANNを学習させると、処方成分や製造プロセスなどの入力因子から、溶出性や安定性などの重要な製剤特性を予測できるようになり、製剤化検討の効率化に役立てることができます。ただし、ANNの予測結果は不安定なことがあるので、その点に注意が必要です。 |
回答:--- |
|
|
|
|
8.60 医薬品の消化管内移動に関係する言葉でハウスキーピングという言葉が出てくるのですが、これはどんなことを意味しているのでしょうか |
|
A: |
絶食時の胃の蠕動運動は、図に示すように4つのモード(空腹期伝播性収縮帯(MMC) と呼ばれています)が約2時間のインターバルで繰り返されています。Phase 1では、ほとんど胃は運動していませんが、Phase IIから徐々に強くなり、Phase IIIは非常に強い収縮で、一気に胃の内容物を十二指腸へと押し出します。この最大の蠕動運動を“胃の内容物を掃除する”ということからHousekeeper wave(参考資料1)と呼んでいます。これが、医薬品とどのように関係しているかですが、例えば、非常に大きな錠剤を服用した場合、Phase I〜IIでは排出されず、Phase IIIでようやく排出されるようなことが起こります。その結果、バイオアベラビリティにも大きな影響が出る可能性があります。また、生物学的同等性試験を絶食下で実施した場合、被験者間のデータのバラツキの原因になる可能性もあります。こうした製剤が胃から排出される時間については、数多くの研究(参考資料1)が行われており、詳細は各論文を読んで頂くと理解することができます。
なお、食事をとると、この運動は大きく変化し、Phase I〜VIのような運動は認められず、一定の運動が継続して発生します。
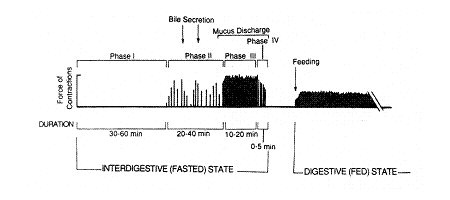
図1:絶食時及び摂食時の胃の運動パターン (参考資料1より引用)
参考資料1: Avraham Yacobi, Eva Halperin-Walega (ed.), Oral Sustained Release Formulations
Design and Evaluation, Pergamon Press, 125-156(1988)
なお、次の報告も参考になります。
J.W. Fara, 剤形の胃排出及び消化管通過における生理学的限界、PHARM TECH JAPAN, 3(5), 15-19(1987) |
回答:--- |
|
|
|
|
8.59 医薬品について、コンパショネートユース(Compassionate Use)という言葉を聞きますが、どういうことなのでしょうか |
|
A: |
コンパショネートユースは“人道的使用”と訳されていますが、公益社団法人日本薬学会の薬学用語解説では、以下のように説明されています。
“基本的に生命に関わる疾患や身体障害を引き起こすおそれのある疾患を有する患者の救済を目的として、代替療法がない等の限定的状況において未承認薬の使用を認める制度。アメリカ、ヨーロッパ(EU)などではすでに導入されており、日本では現在、実施のための検討が行われている。導入に際しては、現行の治験治制度との兼ね合い、対象となる医薬品や患者の選定、未承認薬提供者の限定(製造販売業者、医師、その他)、未承認薬の安定供給の確保、安全性の確保(副作用報告の責務、副作用被害救済制度、感染症被害救済制度の対象の是非などを含む)などが課題となっている。”
なお、コンパショネートユースについて、寺岡らが各国の状況を含め詳細に報告していますので関心のある方は、参考にしてください。
寺岡章雄、津谷喜一郎、未承認薬のコンパショネート使用-日本において患者のアクセスの願いにどうこたえるか-、薬理と治療、38(2)、111-150(2010) |
回答:--- |
|
|
|
|
8.58 ブランドジェネリックとは、通常のジェネリック医薬品と何が違うのですか |
|
A: |
ジェネリック医薬品は新薬(先発医薬品)の特許期間が切れた後、特許を有していた先発メーカー以外の会社(ジェネリックメーカー)が、製造・販売する医薬品を指します。有効成分は同じですが、一般的に添加剤や製剤の大きさ・味・色などが異なります。また、その商品名は有効成分の一般名が使用されたり独自の商品名が付与され、先発医薬品と同じ商品名が使用されることはありません。
一方、ブランドジェネリックは、先発メーカーがジェネリックメーカー(先発メーカーが持つジェネリックメーカーの場合もあります)にブランド名とともに承継し製造・販売するものです。先発メーカーから引き継いでいるので、商品名は新薬時の名称(ブランド)が使用されます。 |
回答:--- |
|
|
|
|
8.57 一般の薬局で薬を買うときに薬剤師から説明を受けましたが、説明が必要になった理由を知りたいのですが |
|
A: |
「薬事法の一部を改正する法律」が2009年6月より施行され、一般薬医薬品の販売制度が見直されることとなりましたが、ここで一般用医薬品をリスクに応じて三つに分類し、そのリスクの程度に応じて専門家が情報提供することとなったからです。
リスクの分類及び情報提供については、以下の表に従います。
医薬品のリスク分類 |
対応する専門家 |
質問がなくても行う情報提供 |
相談があった場合の応答 |
第一類医薬品 |
薬剤師 |
義務 |
義務 |
第二類医薬品 |
薬剤師又は登録販売者 |
努力義務 |
第三類医薬品 |
不要 |
ここで登録販売者とは同制度改正により新たに導入されたもので、一定の実務経験等を有する者が都道府県知事の行う試験に合格し、登録を受けた人のことです。
第一類医薬品は登録販売者では販売できない制度となっていますが、第二類及び第三類医薬品の販売については可能になっています。
リスクの分類については販売されている一般用医薬品の外箱に記載されており、また薬局・薬店ではリスク分類ごとに陳列することとなっています。特に第一類医薬品は販売者から購入者へカウンター越しに医薬品を陳列する方法(Over The Counter)となっています。
更に詳しい情報については 「日本OTC医薬品協会」ホームページにて確認してください。
引用:厚生労働省. “薬事法の一部を改正する法律の概要 |
回答:--- |
|
|
|
|
8.56 「分子標的治療薬」という言葉を最近良く耳にしますが、その特徴について教えてください |
|
A: |
分子標的治療とは、病気の発症や悪化を引き起こしている体内の特定の分子の働きを抑えることで症状を改善させ、さらには治癒を目指す新しい治療法です。科学技術の進歩はヒトの体や病気の仕組みに関し細胞・分子レベルでの解明を可能とし、体内で発病や疾患の進行に重要な働きを果たす分子が特定できるようになりつつあります。分子標的治療薬は、これらの特定の分子の働きだけを特異的に抑えるようデザインされた医薬品です。
一方、国内での分子標的治療は普及から日が浅く、今後未知の副作用が生じることも考えられます。患者さんへの安全性を確保し、効果を最大限に活かすためには、高い専門性と豊富な治療経験を持つ専門医による適正使用と、それを支える最新の副作用情報の収集・報告などの販売企業による万全の安全対策が不可欠です。 |
回答:--- |
|
|
|
|
8.55 特許を維持するための費用は、どのくらい必要なのでしょうか |
|
A: |
特許を取得し、それを維持すためには費用が必要となります。具体的な金額は、特許庁のホームページ( http://www.jpo.go.jp/cgi/link.cgi?url=/tetuzuki/ryoukin/hyou.htm)で見ることが出来ます。簡単に解説しますと、出願費用として15,000円が必要となり、出願審査請求費用として118,000円+(請求項の数x4,000円)が必要となります。こうして特許となった場合、特許が存続する期間(存続期間)中は、維持するための費用として特許料(特許維持年金)を払い続ける必要があります。これを払わない場合には、特許権が消滅してしまう事になります。特許維持年金は、出願審査請求した年度により異なり、以下のようになっています。

|
回答:--- |
|
|
|
|
8.54 特許を出願したいと思いますが、PCT出願をするかと聞かれました。PCT出願というのは、どういうものなのでしょうか |
|
A: |
PCT出願は、海外の国々で自らの発明品に対して特許を取得するための手続きを意味するものです。特許庁のホームページでは、PCT出願に対して、次のように解説されています( http://www.jpo.go.jp/seido/s_tokkyo/kokusai1.htm) 。また、インターネット上では、特許ナビ( http://www.tokkyonavi.com/)にQ&Aの形で、特許に関するいろいろな質問の回答が掲載されていますので参照してください。
(特許庁HPから)
ある発明に対して特許権を付与するか否かの判断は、各国がそれぞれの特許法に基づいて行います。したがって、特定の国で特許を取得するためには、その国に対して直接、特許出願をしなければなりません。しかし、近年は、経済と技術の国際化を背景として、以前にも増して、多くの国で製品を販売したい、模倣品から自社製品を保護したい、などの理由から特許を取りたい国の数が増加する傾向にあります。同時に、そのすべての国に対して個々に特許出願を行うことはとても煩雑になってきました。また、先願主義のもと、発明は、一日も早く出願することが重要です。しかし、出願日を早く確保しようとしても、すべての国に対して同日に、それぞれ異なった言語を用いて異なった出願願書を提出することは、ほぼ不可能といえます。PCT国際出願は、このような煩雑さ、非効率さを改善するために設けられた国際的な特許出願制度です。PCT国際出願では、国際的に統一された出願願書(PCT/RO101)をPCT加盟国である自国の特許庁に対して特許庁が定めた言語(日本国特許庁の場合は日本語若しくは英語)で作成し、1通だけ提出すれば、その時点で有効なすべてのPCT加盟国に対して「国内出願」を出願したことと同じ扱いを受けることができます。つまり、日本人の場合、日本特許庁に対して日本語若しくは英語で作成した国際出願願書を1通だけ提出すれば、それによって国際出願に与えられた国際出願日が、それらすべての国においての「国内出願」の出願日となります。また、PCTは、出願の手続を簡素化するだけでなく、PCT国際出願に独自の制度も用意されています。
たとえば、国際出願をすると、出願した発明に類似する発明が過去に出願された(公知となった)ことがあるかの調査(国際調査)が、すべての国際出願に対して行われます。その際には、その発明が進歩性、新規性など特許取得に必要な要件を備えているか否かについて審査官の見解も作成されます。もちろん、それらの結果は、出願人に提供されますので、出願人は、自分の発明の評価をするための有効な材料として利用することができます。さらに、出願人が希望すれば、特許取得のための要件について予備的な審査(国際予備審査)を受けることもできます(各国が行う特許付与のための審査ではありません)。これらの制度を利用することで、特許取得の可能性を精査し、緻密に厳選した国においてのみ手続きを継続させ、コストの効率化、適正化が可能となります。但し、注意すべき重要な点があります。つまり、PCT国際出願は、あくまで国際的な「出願」手続であるため、国際出願の発明が、特許を取得したい国のそれぞれで特許として認められるかどうかは、最終的には各国特許庁の実体的な審査に委ねられています。
そこで、PCT国際出願の最後の手続は、国際出願を各国の国内手続に継続させるための手続となります。PCT国際出願が国内手続に継続された後は、PCT国際出願もそれぞれの国の国内法令によって処理されます。この「各国の国内手続に継続させる」手続をPCTでは、「国内移行手続」と呼びます。この国内移行手続を行うにあたり、優先日から30ヶ月の期限が満了する前に、権利を取りたいPCT加盟国が認める言語に翻訳した翻訳文をその国の特許庁に提出し、その国が求める場合には手数料を支払う必要があります。 |
回答:--- |
|
|
|
|
8.53 薬物の有害性評価において、TWAという言葉が出てきますが、どういうことを意味しているのでしょうか。OELとは、どう違うのでしょうか |
|
A: |
TWA(Time Weighted Average:時間加重平均)は、ほとんどすべての作業員が通常の1日8時間、週40時間労働とした場合にも健康上の悪影響がないと考えられる薬物の許容濃度で、日本産業衛生学会、米国ACGIHが勧告値を発表しています。
OEL(Occupational Exposure Limit:許容暴露限界)も同様に作業員が通常の1日8時間、週40時間労働とした場合にも健康上の悪影響がないと考えられる薬物の許容濃度で、前臨床試験や臨床試験の薬理データや毒性データをから設定する無毒性量の数値から一般的に下式で求められます。
OEL(mg/m3)=NOAEL × BW / V × MF × S × α
NOAEL:無毒性量(mg/kg/day)
BW:平均体重(kg)
V:吸気量(10m3)
MF:修正係数
S:定常時の調整係数
α:吸収補正係数(1/F) |
回答:--- |
|
|
|
|
8.52 ICタグという言葉を聞きますが、これはどのようなものなのでしょうか。またなぜ話題になっているのでしょうか |
|
A: |
ICタグは、内蔵したメモリのデータを電波(電磁波)により、非接触で読み書きする情報媒体のことで、RFタグ、電波タグ、無線タグ、RFIDタグ等様々な呼び方をされていますが、JISでは、RFタグと規定しています。
電波(電磁波)を用いてデータを非接触で読み書きすることをRFID(Radio Frequency IDentification)と呼びこの場合は、リーダ/ライタも含みます。
RFタグは物に付けて利用しますが、人が持って入退室管理、電子乗車券(Suica・PASMO)、決済用途(Edy)などで使用する「非接触ICカード」も広義のRFIDと言えます。
RFタグの動作は、使用する周波数によって実現方法は異なりますが、RFタグとリーダ/ライタとの間のデータ伝送の原理はほぼ同じで以下の手順となります。
- リーダ/ライタのアンテナから、情報を電波又は磁界に乗せて送信します。
- RFタグのアンテナがリーダ/ライタからの電波又は磁界を受信します。
- 整流(電波の場合)または共振(磁界の場合)により、RFタグのアンテナに電力が発生します。
- 発生した電力により、制御回路、メモリを動作させ、必要な処理を行います。
- RFタグ内のデータを、電波または磁界に乗せてRFタグのアンテナから返信します。
- リーダ/ライタのアンテナで、RFタグからの電波又は磁界を受信します。
- リーダ/ライタの制御部で、電波または磁界から情報を取り出します。
RFタグには、データの書替えができる、汚れ等に強い、ある程度遮蔽物があっても非接触で交信ができる、同時に複数の読取りができるなどの特徴があります。
バーコードに比べ、読み取りが早く1つ1つではなく一括読取りが可能です。また用途に合わせてデータの書き込みが可能などの特徴を持っています。
医薬品の展開例としては、アンプルへの表示などで、バーコードが1管ごとに表示できないような小さな物に対しても、アンプルの底にICタグを付けるなどで解決できる実験例も見たことがあります。
医療現場から考えれば、包装単位だけではなく、如何に調剤単位に情報を表示するかを今後進めて行く必要があると思いますが、ICタグが一つの解決策であることも事実だと思います。
欠点は、まだ単価が高いことと、人間が情報を見られないことだと思います。
RFタグが注目されている理由は、非接触認識や複数同時認識などRFタグの持ついくつかの特性を情報システムに応用することにより、人の手により行われているバーコード運用を含む多くの業務オペレーションを自動化、あるいは簡素化することができるため、莫大なコストを削減できると言われているからです。
さらに、人為的なミスの防止やシステムのリアルタイム性が向上することにより、情報の質が向上するとも言われています。
しかし、課題も多く、コストがまだまだ高い、データが目視できない、周波数によって交信距離や指向性が大きく変わる、水分や金属の影響を受けるなどの問題から限定した用途での採用となっています。RFタグを運用する場合は、その特徴を十分理解して、用途に応じたRFタグを選定することが重要となります。 |
回答:--- |
|
|
|
|
8.51 日本と米国で新薬の承認申請する場合、どのくらいの費用がかかるのでしょうか |
|
A: |
わが国において新薬の承認審査に必要となる費用は、薬事法、薬事法施行令、薬事法関係手数料令などにより決められています。この金額は、厚生労働省のホームページ( http://web.fd-shinsei.go.jp/application/list_drug.html) や独立行政法人医薬品医療機器総合機構などのホームページ( http://www.pmda.go.jp/index.html)などから入手できます。
現時点での費用の詳細は、こうした資料を参照して頂くようにお願いします。なお、2011年の段階で新医薬品製造販売承認(オーファンドラッグを除く)の場合、国と調査機関に支払う手数料は30,881,500円、一般用医薬品の場合、その額は131,600円となっていますが、この金額は承認申請の内容(例えば、新薬、効能・効果追加、新剤形など)により大きく異なります。
一方、米国の場合には1992年新薬審査の迅速化・効率化を目的としたユーザーフィー法(PDUF: Prescription Drug User Fee Act)が施行され、FDAは企業から新薬承認申請が行なわれた場合、審査にかかる費用を徴収する事ができるようになりました。その年のユーザーフィーは、連邦広報(Federal Register)に発表されます。2011年会計年度における臨床試験データの提出を伴う新薬新規(フル)申請の費用は、$1,542,000と報告されています。この金額を$1.00 = 80円として換算しますと、123,360,000円となります。我が国の場合と単純比較は困難ですが、3倍程度費用が必要になります。ユーザーフィー法の詳細については、FDAのホームページ( http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/)や下記の参考資料から情報を得ることが出来ます。
(参考資料)
石居昭夫(著)、FDAの承認審査プロセス 新薬の知識、薬事日報社(2010) |
回答:--- |
|
|
|
|
8.50 中国では漢方薬が中心だと聞きますが、本当でしょうか |
|
A: |
都市部と農村部とでは状況が異なり、都市部では西洋薬がよく使われるが、農村部では中国の伝統医学である中医学で用いる中薬による治療が多い。
(杉田亨:中国中医薬の歴史、製剤機械技術研究会誌、Vol.15 No.4(2006)、25—31 )
なお、中薬では日本のものと同じ漢字名であっても、一般には、使用する生薬量が多く、また同じ生薬名でも異なる植物を用いる場合があり、同じ名前でも中身は異なる場合がほとんどである。加えて、中医理論に基づき、新しい処方(配合薬)が日々、生み出され、販売されている。 |
回答:--- |
|
|
|
|
8.49 漢方と生薬は、どう違うのでしょうか |
|
A: |
生薬は動植物の薬用とする部分、細胞内容物、分泌物、または鉱物などを指し(第16改正日本薬局方生薬総則)、漢方薬は日本の伝統医学のひとつである漢方医学の治療に沿うように生薬を一定の規則により配合したものである(医薬品製造販売指針2010、じほう)。 |
回答:--- |
|
|
|
|
8.48 以前Continuous Processing Systemについての講演がありましたが、メリット・デメリットがあるのでしょうか |
|
A: |
従来から医薬品産業では、バッチプロセスによる生産が主流でしたが、医薬品以外の他の産業、例えば石油、化学、食品では連続生産プロセスが通常採用されています。医薬品産業で連続生産プロセスが採用されなかった背景は、生産量がそれほど多くない医薬品の製造では柔軟性や融通性の観点で適さないと考えられ、また製品の品質やプロセスの機能に対する信頼性が十分でなかったからと思われます。
しかし近年、PATやデザインスペース申請の概念が広まるにつれ、経口剤の製造ラインとしてPATを組み合わせた製造システムが開発され注目を集めています(ConsigmaTM, PHARM TECH JAPAN 2011年5月号参照)。
経口剤の場合を例にメリット・デメリットを説明します。連続生産の場合、造粒工程、乾燥工程、混合工程がスケールの影響を受けないことを示すことが可能なケースがあり、この場合、開発段階で大量の原薬を使用することなく、処方、製造法を短期間に検討することが可能で、かつデザインスペース申請が可能となります。また承認前のバリデーションについてもスケールアップ検討が不要のため、コスト、期間の点でメリットがあると言えます。一方、生産に導入するという観点からは、製造スペースや製造コストを大きく低減できる可能性があります。
一方、課題としては規格外品の排除がバッチプロセスによる生産と比較して困難であり、トラブル発生時の対応が難しいことがあげられます。また品目切り替え時の洗浄にも時間を要するため、生産量の少ない製品には不向きかも知れません。
いずれにしても今後、生産だけでなく、開発段階から適用することにより、PATとデザインスペースの組み合わせによる高度な品質保証を達成する製造法として注目されていくと思われます。 |
回答:--- |
|
|
|
|
8.47 高活性物質(薬物)とか高薬理活性物質といいますが、具体的にどのような薬物のことをいうのでしょうか。またどのようなデータで判断するのでしょうか |
|
A: |
高生理活性物質とは、ある種のステロイド類、抗ガン剤のように、少量で人体に強い薬効を与えるもの又は毒性を有する物質等をいいます。判断基準としては、前臨床試験や臨床試験の薬理データや毒性データをから設定するNOAEL(No Observed Adverse Effect Level:無毒性量)を基本とし、NAOELから算定されるOEL(Occupational Exposure Limit:許容暴露限界)、また無毒性量に安全係数を乗じたADI(Acceptable Daily Intake:一日許容摂取量)などが判断基準として使用されています。(古谷) |
回答:--- |
|
|
|
|
8.46 ヒューマンエラーというのは、どのようなエラーの事を言うのでしょうか。そうしたエラーを効果的に防ぐ手段はあるのでしょうか |
|
A: |
英国の著名な心理学者であるJames Reason は、ヒューマンエラーとは、"計画された行動は適切であるにもかかわらず、計画どおりの行動に失敗すること" と定義しています。この定義からしますと、"偽装、隠ぺい、データ改ざんなど"は、ヒューマンエラーとは言えません。つまり、意図的に生じさせたものはヒューマンエラーとは言わないわけで、それだけヒューマンエラーの発生を防止することは難しいということが出来ます。東京大学の大山正教授は、"人間はもともと間違い(エラー)を犯すもので、完全にこれを無くすことは不可能である。ヒューマンエラーの防止は、まずこの厳しい現実を認めて、それを前提に、ヒューマンエラーに基づく事故の防止策を考える必要がある。"と述べています。こうした事を踏まえ、回答者が考える防止方法としては、産業技術総合研究所の中田亨先生が提案されているように、実施される作業に対して作業者が"四ない"(①興味が湧かない、②自分に関係ない、③自分にはできない、④できてもうれしくない)という気持ちではなく、"四ある"(①面白そうだ・気になる、②自分が使える場面があるぞ、③やればできそうだ、④やって良かった)と言う気持ちを持ってやれるような環境を作る事が大事であると考えています。こうしたヒューマンエラーを防ぐためのいろいろな方法については、数多くの成書(下記参照)がありますので、ぜひ参考にしてください。
(1) 中田亨(著)、"防げ現場のヒューマンエラー—事故を防ぐ3つの力"、日科技連出版社(2010)
(2) 古田一雄、日本原子力学会ヒューマンマシンシステム研究部会(著)、"ヒューマンファクター10の原則—ヒューマンエラーを防ぐ基礎知識と手法"、日科技連出版社(2008) |
回答:--- |
|
|
|
|
8.45 品質工学の中に出てくるS/Nは、処方設計の場などでは、具体的にどのような数値を使って求めればよいのでしょうか |
|
A: |
品質工学でのS/N比は、通信工学の分野で用いられる機能のよさを表す尺度と同様に信号対雑音比、すなわち、有効な情報(Signal、シグナル)/有害な情報(Noise、ノイズ)を表します。よって、S/N比が大きいとは、ばらつきが低く、かつシステムの働きが良いことを示します。品質工学ではシステムの働きを、基本機能 (出力「測定値」をyとし、入力値Mとした時に、y=βM 「βは任意の比例定数)」)で示し、具体的に取り扱う出力値と入力値は、基本機能をどのように定義するかで異なってきます。処方設計の場では、例えば、製剤の溶出性を基本機能とした場合、溶出率(出力y)と溶出時間(入力M)の数値を使用してS/N比を求めます。( 資料参照) |
回答:--- |
|
|
|
|
8.44 最近使えない単位の話を聞きますが、SI単位というのは、何ですか?どうしてSI単位を使うことになったのでしょうか |
|
A: |
"SI"とは、フランス語の"Le Systeme International d'Unites(国際単位系)"の略称です。SI単位は、メートル(m)、キログラム(kg)、秒(s)、アンペア(A)、ケルビン(K)、モル(mol)、カンデラ(Cd)という7つの基本単位、そして、組立単位と補助単位から成ります。
過去には、同一の物理量に対し、様々な単位が混用されてきました。しかし、換算の必要があり効率的でないため、世界で共通の単位を使用しようと、1954年に行われた第10回国際度量衡総会で、SI単位の導入が決定されました。日本では、1993年にSI単位系を採用した計量法が施行されました。非SI単位は、安全上の理由から、最長で1999年までの猶予期間が設けられ、その後、公的な場での使用ができなくなりました。 |
回答:国際委員会 |
|
|
|
|
8.43 微粉と言いますが、どのくらいの物をいうのでしょうか |
|
A: |
どのくらいの大きさの粉が微粉なのか、明確な定義はありません。粉粒体を大きい方から粒体、粗粉体、微粉体、超微粉体の4つに分類した場合、下表に示すように様々な定義がされています。また、粉粒体には粒度分布があります。取り扱う粉粒体の粒度分布によっては、仮に粗粉体の範囲であっても微粉と見なす場合もあり、相対的にとらえることが必要です。
| 区分 |
文献1 |
文献2 |
文献3 |
| 粒体 |
10 mm以上 |
1 mm以上 |
数10 m以上 |
| 粗粉体 |
100 μm以上10 mm未満 |
10 μm以上1 mm未満 |
約3 μm〜数10 μm |
| 微粉体 |
100 nm以上100 m未満 |
10 nm以上10 m未満 |
約300 nm〜約3 μm |
| 超微粉体 |
1 nm以上100 nm未満 |
0.1 nm以上10 nm未満 |
1 nm以上〜約300 nm |
参考文献
- 川北公夫、小石真純、種谷真一共著 粉体工学(基礎編)(1974)槇書店
- 川北公夫著 粉粒体のトラブル対策(1980)日刊工業新聞社
- 神保元二著 粉体の科学(1985)講談社
|
回答:国際委員会 |
|
|
|
|
8.42 ゲノム創薬というのは、どのようなことをいうのでしょうか |
|
A: |
従来の創薬手法は疾病の病態研究から受容体拮抗や酵素阻害活性を指標に数万種類の化合物をスクリーニングする偶然性に頼っていました。生物の有する遺伝情報の全体をゲノムと称しますが、近年の分子生物学や情報科学(バイオインフォマティクス)の進展によりヒトゲノムの解読が進み、癌や糖尿病、高血圧など数多くの疾患に遺伝子が関連していることが明らかになってきました。これらのゲノム情報を活用することにより、疾病関連遺伝子及びタンパク質を特定し、医薬品の標的を明確にする論理的・効率的な創薬手法をゲノム創薬といいます。更に病気の原因や薬物応答の個人差の原因となる遺伝子をみつけ、より効果が高く、副作用の少ない医薬品の創生が可能となり、個人の特性を考慮したゲノム創薬によるテーラーメード医療が期待されます。 |
回答:国際委員会 |
|
|
|
|
8.41 医薬品の価格の話を聞くとDRG(Diagnosis Related Group)という言葉が出てきますが、これはどのようなことをいうのでしょうか |
|
A: |
DRGは疾患別関連群と訳され、PPS(Prospective Payment System)包括支払方式と組み合わせてDRG/PPS、すなわち、診断に基づいて患者を分類(DRG)し、疾患毎に定められた定額の医療コストが医療機関に支払われるという仕組みを意味します。DRG/PPSは、1983年米国において採用されましたが、日本ではこのDRGの考え方を改良してDPC (Diagnosis Procedure Combination;診断群分類)が提案され、2003年に国内でDPCに基づいた診断群分類包括評価を用いた入院医療費の定額支払い制度が開始されました。
これまでは、治療にどれだけの費用が掛かったかで報酬が決まっていましが(出来高払い)
、定額支払い制度は、患者が何の病気であったか(診断群分類)によって診療報酬が一律に決まる制度です。
これまでの出来高払いでは、効果的な医療行為を行い最短期間で回復させる治療に比べ、回復を長引かせ、多くの医療行為を行なった医療者への支払いが増えると言う矛盾が起き得ます。一方、DPCを用いた定額支払い制度であれば、診断結果によりDPCで分類された診療報酬が決められていますから、実際に掛かった医療費は後から経費として差し引かれ、残った額は利益となります。つまり、回復への最短治療を行った医療者は、利益を得ることができ、患者は早期回復できるという利益を得ます。また、従来の診療では採算割れの傾向が強かった急性期病院は経営的安定が確保できることや、医療費抑制の効果なども期待されています。 |
回答:国際委員会 |
|
|
|
|
8.40 米国の医療制度の話を聞くとマネージドケアやHMOという言葉が出てきますが、これはどのようなことをいうのでしょうか |
|
A: |
米国では国民皆保険制度が採用されておらず、メディケア/メディケイドの公的医療保険を除くと民間保険が採用されています。大企業では、従業員確保の手段として1980年代まで医療保険を提供していましたが、医療費の高騰によりこうしたシステムは崩れ、従業員も何らかの負担をする状況となっています。米国では医療機関が行っている医療行為に対する費用を医療機関自ら設定することができるため、人気のある病院や実績のあるところでは非常に高額の医療費が必要となります。例えば、日本では出産費用として30万円程度ですが、米国では150万円にもなるといわれています。従って、当然のことながら民間保険に加入することになるのですが、このように費用が天井知らずの状況では、保険会社はすぐに破綻してしまうか、とんでもなく高い保険料となってしまいます(無保険者が多いという理由もこうしたところに一因があります)。そのため多くの民間医療保険には、マネージドケア(医療内容に医師以外の保険者が介入し、被保険者が受ける医療サービスに何らかの管理や制限をくわえ、医療サービスの提供に関してコスト抑制意識を高めるとともに効率的な医療を提供する仕組み)が取り入れられています。HMO(Health Maintenance Organization:健康維持組織)は、年間一定の医療保険料を支払えば指定された病院や医師の診療や薬剤給付が受けられる会員制の組織で、医療費の増加に歯止めをかけるために80年代後半から普及したマネージドケア(管理医療)の代表格です。会員は一定額を支払えば一定水準の医療サービスを受けられます。会員が健康なほどHMOと医療機関は利益を上げることが出来、HMOは会員の健康維持・疾病予防に力を入れています。HMOは利益確保を目指し、なるべくコストの安い病院や医師と契約し、新薬やブランド薬の使用を控え薬剤費も削減しています。簡単な病気なら薬剤や医療用品を自宅に送るだけで済ませるほか、入院期間の決定権もHMOが握っています。患者負担は出来高払いの従来型保険に比べ10〜40%少ないといわれています。代表的なHMOとして、ユナイテッド・ヘルスケア、エトナ、ヒューメナカイザー・パーマネンテ、オックスフォード・ヘルス、ヘルス・プラン、パシフィケア・ヘルス・システムなどがあります(参考資料:月刊医療情報1998年11月号)。マネージドケアには、この他にもPPO(Prefered Provider Organization)やPOS (Point of Service)などがあります。
(参考書)
- 柿原浩明、入門医療経済学、日本評論社(2004)
- E.M.コラッサ、米国における医療用医薬品価格戦略-日米の比較も含め、(訳)佐賀国一、(株)じほう、2001
- http://www.kaigo.gr.jp/lab/usan.htm
(この他にも、多くの関連書籍が販売されています。また、ネットからも情報を取ることができます。) |
回答:国際委員会 |
|
|
|
|
8.39 希少医薬品というのは、どのような医薬品をいうのでしょうか |
|
A: |
希少医薬品(オーファンドラッグ)とは、次の3つ定義 *注1に適合する医薬品のことを意味します。
- 国内の患者数が5万人未満の重篤な疾患に使用される医薬品
- 適切な代替医薬品がない、あるいは既存の医薬品の有効性や安全性が低いなど、医療上の必要性が高い医薬品
- 開発が成功する可能性の高い医薬品
製薬企業にとり、患者数が非常に少ない疾患などに使用される医薬品の開発は、採算の面からあまり魅力のあるものとはいえません。しかし、患者さんにとっては生命にかかわる重要な問題であり、国は1993年改正薬事法の中で「医療上特にその必要性が高い医薬品及び医療用具の研究開発促進のために必要な処置を講ずること」を明記し、「希少疾病用医薬品・希少疾病用医療用具の研究開発促進制度」を設けて、こうした希少医薬品の開発援助を行っています *注2。英語でオーファンとは「孤児」といった意味がありますが、どの企業もあまり開発したがらない医薬品、孤児のような医薬品であることからオーファンドラッグと呼ばれるようになったとの話があります。
*注1:この定義は国により異なっています。
*注2: http://www.nibio.go.jp/shinko/orphan.html |
回答:国際委員会 |
|
|
|
|
8.38 最近 Counterfeitという言葉を目にしますが。これは何ですか。なぜ注目されているのですか |
|
A: |
Counterfeitは「偽薬医薬品」のことで、その正体や出所・起源を故意に偽った医薬品でのことです。その種類は、有効成分が表示通り含まれているもの、有効成分の含有量が異なるもの、有効成分が含まれていないもの、本来の主薬とは異成分を含んだものなど、いろいろなCounterfeitがあります。こうした偽物が出回ることにより、製薬企業の本来得られるべき利益が損なわれるだけでなく、そうした偽薬を服用した患者の生命に調節影響を及ぼす可能性があります。また、こうした医薬品が犯罪に結びついている可能性も指摘されています。偽薬の問題は、例えば発展途上国ではオリジナル医薬品の値段が高く入手できず、そのためこうした偽薬が出回りやすい環境があり、単に区別がつけばよい、出来ないようにすればよい、といった技術的な問題以外の問題も含まれていることに注意する必要があります。先進国におけるCounterfeitの割合は1%未満と非常に少ないのですが、アフリカ諸国やラテンアメリカの一部では、これよりはるかに高い割合となっています。FDAでは、2009年「Incorporation of Physical-Chemical Identifiers into Solid Oral Dosage Form Drug Products for Anticounterfeiting」なるドラフトガイダンスを発行し、偽薬の区別および防止に対する取り組みを始めました。
(参考資料)
(1) 製剤機械技術研究会 平成22年度 特別講演会 講演要旨集 pp1-29 (2010)
(2) WHO, Counterfeit Drugs, Guidelines for the development of measures to combat counterfeit drugs, WHO/EDM/QSM/99.1 (1999) |
回答:国際委員会 |
|
|
|
|
8.37 バイオシミラーとは何ですか |
|
A: |
バイオテクノロジー技術を用いて微生物や培養細胞により生産される抗体、遺伝子組み換えタンパク質やポリペプチド及びそれらの誘導体を構成する医薬品を一般的にバイオ医薬品と総称します。バイオシミラー(バイオ後続品ともいう)とは既に新有効成分含有医薬品として承認された先行バイオ医薬品と同等/同質の品質、安全性、有効性を有する医薬品として、異なる製造販売業者により開発される医薬品を指します。バイオシミラーは品質、安全性、有効性について、先行品との比較から得られた同等性/同質性を示すデータ等に基づき開発をします(参照)。 バイオシミラーの同等性/同質性評価においてはICHガイドライン:「生物薬品(バイオテクノロジー応用医薬品/生物起源由来医薬品)の製造工程の変更にともなう同等性/同質性評価」に記載されているコンセプトにもとづいた適切な試験の実施が必要です。低分子医薬品では、先発品の有効成分と"同一"の化学構造をもつ成分を後発品として供給することができます。一方、タンパク質、ポリペプチドなど生物薬品では、もともと分子構造が不均一なものが生産される可能性が本質的に存在します。そのため、後発品の有効成分が先発品の有効成分とまったく同一とすることは困難であることから、"シミラー"と表現されます。
(参照)平成21年3月4日 薬食審査発第0304007号 厚生労働省医
薬食品局審査管理課長通知「バイオ後続品の品質・安全性・有効性確保のための指針」 |
回答:国際委員会 |
|
|
|
|
8.36 実験計画法と品質工学は、どう違うのでしょうか |
|
A: |
実験計画法と品質工学の違いについては、いろいろなところで議論されています。どちらも直交表を使用することなどから、その違いについてなかなか分かりにくいと思います。ここに記載します回答も、回答者の理解であって、これが100%正しいと言い切れない部分があります。参考となる書籍などを紹介しますので参照してください。
- 実験計画法は、どの因子が効果(特性)に影響を及ぼしているか、因子間で交互作用があるか、などの原因を追究することに主眼があります。つまり、因子の値の変化に対して、評価している特性値がどのように変化するかを調べ、有意差検定や寄与率で効果があったかどうか、或いはどの程度の影響があったかなどの評価が行われます。ここにおけるバラツキは偶然誤差であり、因子の効果がこの誤差に対して評価されます。因子間の交互作用の影 響を統計学的に見積もることができるのも実験計画法の特徴の1つです。
- 品質工学では、その製品に求められている機能が安定して得られるかどうかに主眼が置かれています。そのため評価の尺度としてSN比(品質工学の場合、期待している特性によりSN比の計算方法が異なります。例えば、望大特性の場合には平均値と標準偏差の比が使われます。)や感度が使用されることになります。そして、このSN比が大きくなるような条件を探すことになります。この場合、バラツキは使用環境や劣化により必然的に変わるものとしてとらえることになります。因子間の交互作用を評価する列はなく、交互作用は全ての列に 等分に現れます。品質工学による取り組みを行うことで、コスト削減や経費低減や開発期間短縮などを図ることができるとされています。
このように両者には違いがありますので、目的に応じて使い分けるのが良いでしょう。製剤開発の段階では、どちらかというと実験計画法の方が良いと思いますが、いざ開発が進み生産条件などを設定する場合には、品質工学による手法が有効かもしれません。
(参考資料)
(1)田口玄一、<復刻版>第3版 実験計画法 上(および下)、丸善(株)、2010
(2)矢野宏、品質工学入門 —技術を変革する新しい考え方、日本規格協会、1995
(3) http://www.qes.gr.jp/introduction/index.htm
(4) http://www.hinkai.com/
なお、実験計画法や品質工学に関する書籍は、数多く出版されています。しかし、数学的な取扱いが多いものや、ある程度基本的なことを理解していないと説明を理解できないものなど、その内容はいろいろです。自分にあった本を選択する事が必要でしょう。 |
回答:国際委員会 |
|
|
|
|
8.35 オーダーメイド医療とテーラーメイド医療は、どう違うのでしょうか |
|
A: |
"オーダーメイド医療"も"テーラーメイド医療"も、「個の医療」あるいは「個人の特性に合わせた医療」という同じ意味で用いられています。これは、個人の特性に応じた治療を行おうというもので、遺伝子レベルから個々の疾病の状態までを見据えたうえで、治療法が決められます。一方、この言葉の対極にある言葉が、"レディーメイド医療"といわれるものです。レディーメイド医療の場合、治療は個人の状態を必ずしも十分に考慮せず、疾病に基づいた画一的な治療が行われます。近年、医療科学の進歩とともに、医療の現場は、"オーダーメイド医療"(テーラーメイド医療)の方向に向かっています。
なお、"オーダーメイド医療"と"テーラーメイド医療"を違った意味で使用する人もいます。その場合、"テーラーメイド医療"は、遺伝子治療に着目した個の医療に、"オーダーメイド医療"は、従来の医療を個々の患者の状態に合わせて適用するような個の医療に使用されているようです。 |
回答:国際委員会 |
|
|
|
|
8.34 会社情報の中でCEOという言葉が出てきますが、社長(President)と、どう違うのでしょうか |
|
A: |
CEOとはChief Executive Officerの略であり、最高経営責任者と訳されます。「会社経営」から見た役職です。これは米国の会社経営制度における役員、執行役員、執行役のトップに立つ役職です。取締役トップの会長(Chairman)や社長(President)とは異なりますが、実際には会長がCEOを兼ねることが多いようです。
一方、「社長」とは会社などにおける最高責任者の「呼称」です。会社法では会社の代表権があるのは取締役か、代表取締役であり委員会設置会社(米国会社経営制度に近いシステム)では代表執行役となっています。「社長」や「会長」などの役職は会社法の規定では存在せず、会社職制による呼称です。
法の規定では以上のように明確ですが、一般的には「CEO」と「社長」は同義で使用されていることが多いようです。 |
回答:国際委員会 |
|
|
|
|
8.33 お医者さんからいろいろな薬をもらいました。この薬に関して、いろいろな情報を知りたいのですが、ネット上などでは、どこを調べたらよいでしょうか |
|
A: |
最近は薬を処方するときに、病院に調剤室あるいは調剤薬局の薬剤師さんがそれぞれの薬の薬効などについて詳しく説明すると同時に薬袋にそれぞれの名称、服用数などを直接印刷したり、あるいは薬の写真も合わせて印刷したものを渡されていると思います。こうした説明等を受けた上で、更に情報をお知りになりたい場合は各製薬会社のホームページに「薬の相談室(各社で名称は異なると思います)」がありますので、予め薬剤師さんにそれぞれの薬の会社名をお聞きになってから製薬会社に問い合わせをすれば情報を得ることが出来ます。なおインターネットで簡便に調べることも出来ますが製薬会社に問い合わせを行い、専門家による対応をしてもらうことが情報の交錯などを避ける意味でも重要であると思います。 |
回答:国際委員会 |
|
|
|
|
8.32 最近、薬学部6年制が話題になっていますが、その中で共用試験とか、CBT或いはOSCEということを耳にしますが、これは何のことでしょうか |
|
A: |
薬学部では、医療技術の高度化、医薬分業などの進展に伴い、高い資質を有する薬剤師養成を目的とし、平成18年4月入学生から修業年限が6年に延長されました。そして、こうした変更に伴い、カリキュラムや実務実習の変更が行われることになりました。特に、5年次行われる実務実習は、従来の見学型の実習から実際の薬剤師業務を経験する体験型・参加型の実習に変更となりました。また、実習期間もこれまでは0.5〜1月程度であったものが、病院/薬局の実習を合わせ6ヶ月(含む事前実習)もの長い期間となりました。このため、実際に薬学生が、この参加・体験型実務実習を行うだけの資質有しているかどうかを確認することが必要となり、そのための方法として行われるものが、共用試験といわれているものです。共用試験には、CBT(Computer-Based Testing)とOSCE(Objective Structured Clinical Examination)の2つの試験があります。CBTは、薬学生として必要な専門知識、実習を行う前に最低限必要な知識・問題解決能力などをパソコン上で出題される問題(選択肢問題 310問)に、1問1分で回答する形で実施される客観試験です。一方、OSCEは、薬剤師としてのコミュニケーション技術や調剤・薬剤監査などの技能や態度を実際に与えられた課題を実施することで確認する実技試験、模擬患者が参画したシュミレーションテストとなっています。(1)患者・来客者対応、(2)薬剤調製、(3)調剤監査、(4)無菌操作の実践、(5)情報提供の範囲の中から6題出題されることになっています。詳細は、薬学共用試験センターのHP( http://pc5.phcat-unet.ocn.ne.jp/center.html)などを見てください。
参考: http://pc5.phcat-unet.ocn.ne.jp/about.html |
回答:国際委員会 |
|
|
|
|
8.31 QOLを数値にして、具体的に評価する方法はあるのでしょうか |
|
A: |
QOL(Quality of Life)は、「人が充実感や満足感を持って日常生活を送ることが出来ること」(QOL研究会HPより)を意味している。この他にも、「薬の効果や医療行為の効果を、患者の立場から評価するもの」と考えることもできます。QOLは、言葉としては美しいのですが、概念としては暖味で、単純な言葉で定義することが難しい概念です。QOLを数値化する方法としては、QOLを評価するための尺度が記載された質問用紙に回答してもらうデータ収集方法が一般的です。この評価法(尺度)としては、以下の3つがあります。
- 包括的評価(generic instrument)
あらゆる疾患や病態に共通に利用することを目的とした尺度
(例)SIP(Sick Impact Profile), NHP(Nottingham Health Profile)
- 特異的尺度(specific instrument)
特定の疾患や病態に対応した尺度
(例)癌患者薬物療法のQOL
- 選好に基づく尺度(preference-based measure)
0—1の間隔尺度で表されたQOLスコアを測定する
(例)評点尺度法、時間得失法、標準的賭け法
また、このような数値化に関しては、次のように分類することもできます。

この他にも、健康状態を数値化したものとして、代表的なものとして「質調整生存年」があります。これは、健康状態を数値化し、その変化を示す曲線下の面積で表すものです。
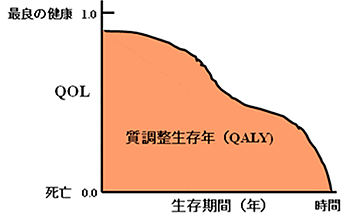
こうしたQOLの評価の詳細は専門書をお読みください。下に、参考となる書籍などをあげておきました。
QOL評価の具体的な例としては、藤野ら(藤野志朗他、薬理と治療、20(12), 311-333(1992))によるエストラジオールによる更年期障害症状改善例があります。この他にも、非常に多くの報告がありますので、興味のある方は参照してください。
参考:
- 臨床のためのQOL評価ハンドブック、(編)池上直巳、福原俊一、下妻晃二郎、池田俊也、医学書院(2001)
- QOL研究会HP(http://qol.jp/)
|
回答:国際委員会 |
|
|
|
|
8.30 医薬品の価値と言いますが、医薬品の価値というのはどうやって決まるものなのでしょうか |
|
A: |
医薬品の価値は、もちろん疾病を治癒できるかどうかにあります。完全な治癒をもたらすような医薬品で、類似の医薬品がないようなものは価値が高いということができます。しかし、医薬品の中には、効果や手術など医療行為や患者のQOLに対する影響が、似たようなもの数多くあります。近年、新薬を開発する場合、こうした医薬品の効果やQOLに対する寄与などを経済的な観点から評価する薬剤経済学という分野が注目されています。
つまり、医薬品の価値を経済的な面から評価しようというものです。詳細は専門書を参照して頂きたいのですが、例えば この医薬品を使えば1年寿命が延びるのですが、100万円かかります。一方、別な医薬品の場合、寿命は半年しか延びませんが、10万円しかかかりません。医薬品として、どちらの医薬品が、より価値が高いといえますか、という問題です。もちろん、この場合、個人の価値観が入りますので、こうした面の数値化も必要となります。具体的な方法としては、下に様な4つの方法があります。面白い例としては、禁煙パッチの薬剤経済的な評価があり、使用しない場合と使用した場合の医療費や寿命の検討結果があります(藤野志朗他、臨床医薬、10(7), 1477-1492(1994))。
薬剤経済学的な評価方法
- 費用最小化分析(Cost-Minimization Analysis)(CMA)
効果が全く同じ複数の診断・治療法について、その費用を比較
- 費用—効果分析(Cost-Effectiveness Analysis)(CEA)
同一の尺度を用いて複数の医療行為の効果を定量的に評価し、その費用を比較
- 費用—効用分析(Cost-Utility Analysis)(CUA)
全く異なった種類の診断・治療法であっても、その効果を共通尺度で表すことにより比較 → 質調整生存年
- 費用—便益分析(Cost-Benefit Analysis)(CBA)
効果を全て金銭価値に置き換えて評価
参考書:
- 久繁哲徳、西村周三 (訳) 臨床経済学、篠原出版、1990
- 川渕孝一、わかりやすい医療経済学、(株)日本看護協会出版会、2001
- 坂巻弘之、やさしく学ぶ薬剤経済学、(株)じほう、2003
- 池田俊也、坂巻弘之(監訳) 実践 薬剤経済学、(株)じほう、2000
- 柿原浩明、入門医療経済学、日本評論社、2004
- 藤野志朗、医療と医薬品の経済分析、東洋経済新報社、1999
|
回答:国際委員会 |
|
|
|
|
8.29 最近、抗体医薬品が注目されています。
(1) この原薬に相当するものは、どのような物性ですか?粉末あるいは溶液状態ですか? |
|
A: |
抗体医薬品原薬の物性は基本的にはタンパク質溶液です。タンパク質濃度は溶解液の濃度にもよりますが通常、タンパクを安定させ克つ生体に投与しても安全であることが確認されている緩衝液が使用されます。保存は溶液のまま低温保存、凍結保存、あるいは凍結乾燥粉末の形態ですが当該タンパクの性状、溶液での安定性(一般的にタンパク質は不安定)、運搬や使用の際の利便性によりその形態が選択されます。 |
(2) 抗体医薬品としての製剤は、リポソームに抗体付着させて注射剤として投与するものですか?内服固形剤としての開発は困難なのでしょうか? |
|
A: |
通常、タンパク質は生体内でプロテアーゼにより速やかに分解されるため単独で投与するとその薬効は減弱します。そこで製剤の安定化を図り標的部位への到達、体内動態の最適化を図るためにDDS製剤として剤型が工夫されます。リポソームはその一例です。
タンパク質はその分子量が大きいため(通常10KDa以上)経口投与後、消化器官からの吸収は困難です。そのため通常は静注あるいは上述のようなDDS製剤として投与される必要があります。しかし内服固形剤の開発も試みられています。 |
回答:国際委員会 |
|
|
|
|
8.28 後発医薬品に関して、米国ではハッチ・ワックスマン法というのがあると聞きましたが、これはどのような法律のことでしょうか |
|
A: |
ハッチ・ワックスマン法は、正式には“The Drug Price Competition and Patent Term Restoration Act of 1984”(医薬品の価格競争と特許期間回復法)のことで、起案者である上院議員Hatch氏と下院議員Waxman氏の名前からHatch/Waxman法と呼ばれているものです。1984年レーガン大統領により署名・発行されました。
この法律は、次の2つの問題を解決するために出されたものです。
- 新薬の開発・承認に時間がかかり、上市できてもすぐに特許期間が切れてしまう。
- 後発医薬品の承認プロセスが複雑過ぎ、高額の開発費と各種試験が必要であった。
ハッチ・ワックスマン法では、生物学的同等性試験で後発医薬品の申請を可能とし新規の治験は必要としないこと、先発品の特許期間中に後発品の開発を可能とすること、新薬にデータ保護期間(5年間)や一変申請に対するデータ保護期間(3年間)を認めること、試験及び審査期間中に消失した特許期間の回復を認める(最大5年間)などが含まれています。このハッチ・ワックスマン法は、先発メーカー、後発メーカー両者にメリットをもたらし、一方で後発医薬品の使用促進が大きく進展しました。詳細は、資料を参照してください。
参考資料:渡辺敏一著、Balance of Power - ジェネリックvs. 先発企業、pp54 - 58、株式会社医療経済社(2006年) |
回答:国際委員会 |
|
|
|
|
8.27 結晶多形ができやすい分子はあるのでしょうか |
|
A: |
分子構造と結晶多形の発現の関係については計算化学により分子構造から生じ得る結晶形を予測しようとする試みが、古くからなされています。しかし、アルゴリズムの改良や近年の飛躍的なコンピューターの処理能力の向上にもかかわらず、部分的な成功例があるにすぎません。この理由は、主に結晶内にパッキングされた分子間相互作用を正確に推測することが、困難なためです。
このように、結晶多形のできやすさを分子の構造から予測することは現時点では困難です。従って、“結晶多形ができやすい分子はあるのか?”というご質問への回答は、“現時点ではわからない”となります。
しかし、一般論として結晶中の分子間に働く非共有結合の相互作用のうち最も重要なものは水素結合です。このため、同種の他分子と複数の水素結合様式の組み合わせを形成することができる分子構造、特に水素結合の形成の仕方により、分子が異なるコンフォメーションを取りえるような構造の分子は結晶多形を形成する可能性が高いと言えるでしょう。 |
回答:国際委員会 |
|
|
|
|
8.26 結晶多形を簡単に作る方法はありますか |
|
A: |
ご質問の内容を、“医薬品またはその候補化合物の準安定結晶を取り出す簡単な方法”に関するものとしてお答えします。
結論から言いますと、そのような汎用性のある簡単な方法は残念ながらありません。
医薬品またはその候補化合物の場合、大部分の化合物で結晶多形また擬似結晶多形(ある種の水和物等)が存在すると言われています。医薬品開発において開発のできるだけ早い段階で、出現する可能性のある結晶形を評価しておくことは非常に重要です。
しかし、晶析操作時の、1)核発生、2)結晶成長、3)転移速度の3つの要素が準安定形結晶にとって有利な場合にはじめて、準安定な結晶形が取り出せることになります。これらの3つの過程はそれぞれが、多くのパラメーターにより影響を受け、現象は複雑です。このためにできるだけ多くの条件で結晶化を試みる網羅的結晶多形スクリーニングの受託会社が存在し、非常に高価なハイスループット晶析スクリーニング装置が市販されています。
こうした網羅的スクリーニングでは出来るだけ多くの結晶形を析出させるために、溶媒や冷却晶析における冷却速度を変える、あるいは冷却法以外の晶析方法(蒸発や貧溶媒の添加等)に変えたりします。
核発生に関してはOstwald の段階則により、結晶化の駆動力が大きい、つまり過飽和度が大きく結晶化の速度が大きいほど準安定晶が得られやすいといわれています。一般論としては、冷却晶析では冷却速度が大きいほど、準安定結晶が取り出せる可能性が高いと言えます。 |
回答:国際委員会 |
|
|
|
|
8.25 新薬の成功確率は、化合物1万個に1個程度と聞きますが、臨床試験に入ったものの成功確率はどの程度なのでしょうか |
|
A: |
|
回答:国際委員会 |
|
|
|
|
8.24 プラセボ(Placebo)効果に対して、ノシーボ(Nocebo)効果という言葉があると聞きますが、これはどんなことなのでしょうか |
|
A: |
プラセボ効果については、このQ&Aコーナーで既に取り上げられています。 薬効成分が入っていない製剤でも30%程度は効果が出るというのがプラセボ反応ですが、一般的にプラセボ効果は、効果が出る、つまり患者にとって、あるいはその疾患にとってポジティブな反応を意味しています。一方、ノシーボ効果というのは、逆に患者に対して、あるいはその疾患に対して起こるネガティブな効果を意味しています。ノシーボ効果の例を出す前に、ここで注意しておきたいことは、プラセボ製剤を投与した時に起こる反応だけがプラセボ効果ではないということです。たとえば、お医者さんが、“あなたの症状は軽く、すぐに良くなりますよ”といった場合、それだけで症状が良くなることがあります。この反応も、一種のプラセボ効果といえるものです。これとは逆にノシーボ反応は、“ネガティブな期待をいだけば、健康に悪い影響を与える可能性が高まること”ということで、 たとえば、胃が痛いときに、お医者さんに“単なる食べすぎで何も心配要りません。明日には良くなっていますよ。”といわれると、プラセボ反応が起こるかもしれませんが、逆に“そりゃ胃癌かもしれません。明日精密検査をして見ましょう”といわれると、本当はたいしたことがなくても症状は悪化することがあります。後者は、まさにノシーボ効果の例です。プラセボ効果やノシーボ効果については、下記の参考書に詳しく紹介されています。
参考書:ハワード・ブローディ著、伊藤はるみ訳 “プラシーボの治癒力 心がつくる体内万能薬” 日本教文社(2004) |
回答:国際委員会 |
|
|
|
|
8.23 医薬品のライフサイクルとは、どんなことをいうのでしょうか |
|
A: |
常医薬品のライフサイクルという場合、製品が承認され市場に出てから、製造中止・販売終了までの売上の推移のことを言います。この推移は、一般的に図のような曲線を描きます。この曲線には、市場に導入した時期 ⇒ 成長期 ⇒ 成熟期 ⇒ 衰退期 と、大きく4つのフェーズがあり、「ライフサイクル曲線」とも呼ばれます。人の一生における活動エネルギーも、このような曲線をたどります。この曲線下の面積は、その製品の総売り上げとなります。ライフサイクルマネジメントとは、この面を最大にするような取り組みということが出来ます。なお、「製剤開発に関するガイドライン」(薬食審査発第0901001号)では、“初期開発から市販を経て製造販売中止に至るまでの製品寿命の全過程”と定義されています。
参考資料: http://www.chikennavi.net/word/druglifecyclemanagement.htm
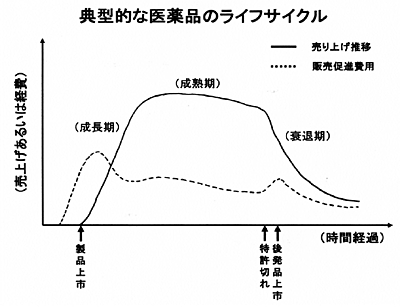 図:医療用医薬品のライフサイクル |
回答:国際委員会 |
|
|
|
|
8.22 Pharmacovigilanceに関して簡単に教えてください |
|
A: |
Pharmacovigilanceは、“医薬品安全性監視”と訳されています。ICHの2E2におけるガイダンス文書“Pharmacovigilance Planning”(薬食審査発第0916001)*では、次のように述べられています。
“医薬品の承認決定は、医薬品の添付文書に特定された条件下でベネフィットとリスクのバランスが満足すべきものであることに基づいて行われる。この決定は、承認時点における入手可能な情報に基づいて行われる。医薬品の安全性プロフィールに関連する知見は、患者背景の拡大及び使用症例数の増加に伴い、時間の経過とともに変化する。特に市販後早期においては、臨床試験とは異なる状況下で使用され、比較的短期間にはるかに大きな集団に使用される可能性がある。
医薬品が上市されると新たな情報が生まれ、それは医薬品のベネフィット又はリスクに影響し得る。これらの情報の評価は、規制当局との協議の下に行われる継続的なプロセスであるべきである。医薬品安全性監視活動を通じて生み出された情報の詳細な評価は、すべての医薬品にとって、その安全な使用を保証するために必須である。医薬品使用者への時機を得た情報のフィードバックを可能にする効果的な医薬品安全性監視を通じて、患者のリスクを低減することによってベネフィットとリスクのバランスを改善することができる。“
つまり、承認段階における医薬品の安全性評価は限られたものであり、上市された後も継続的に評価を行うことが必要であり、それにより医薬品の患者に対する真の安全性を評価することが可能となる。Pharmacovigilanceとは、こうした医薬品の安全性に関する評価と監視のことである。
参考資料: http://www.pmda.go.jp/ich/e/e2e_05_9_16.pdf |
回答:国際委員会 |
|
|
|
|
8.21 薬学部が6年制になりました。4年生の薬学部もあると聞きます。何がどう替わったのですかか |
|
A: |
薬学教育・薬剤師養成教育の歴史に残る、教育年限4年から6年への延長が平成18年4月に実施されました。これは平成16年5月に国会において学校教育法が全会一致で改正されたことによるものであり、その第55条②は「(略)薬学を履修する課程のうち臨床に係る実践的な能力を培うことを主たる目的とするもの(中略)の修業年限は、六年とする。」となりました。
(1)なぜ「薬学」の修業年限が六年ではなく、長い修飾文が付くの?(2)なんで薬学がこの時期に6年になるの?との質問によく遭遇します。最初の疑問に対しての回答は、明治以来の日本の国策・薬学教育の歴史にまで遡らなければなりません。明治・大正時代の「薬学」の位置づけは主に富国強兵の中、主要医薬品の国内生産にありました。およそ80年前、1918年第一次大戦の終結の年、千葉医科大学亥鼻地区に薬学新校舎が完成した折、祝辞に「欧州ノ天地煙砲ニ包マルルヤ、医薬品ノ輸入ハ途絶シ、(中略)此好機ニ於イテ三輪校長ガ薬学教室ノ改築ヲ文部省ニ要求セラレタルハ洵ニ當ヲ得タルモノ‐‐」が寄せられていることからも理解できます。現在も、医療の担い手たる優れた薬剤師を養成することと合わせて、医薬品産業を支える技術者・科学者・開発担当者などを輩出すること、保健衛生のスペシャリストを養成することはわが国の薬学に課せられた使命といえます。米国では薬学部卒業生の90%以上が病院・薬局で働くのに対して、日本でのその割合は40%に満たないのが実情です。こうした背景から「主に薬剤師養成を目的とする6年コースと、技術者・科学者などを養成する4年コースの並立」という他の国に例の無いユニークな薬学部のシステムが誕生することとなりました。この制度の長所には、「現状のような全学生ではなく、真に薬剤師免許を必要とする高いモチベイションを有する学生に薬剤師教育が施せる。」「総合的科学である薬学を学ばせるにあたって、目標に応じたコンデンスした教育が可能となる。」などがあります。逆にこの制度が定着するまでは同じ薬学部でも薬剤師になれたり、なれなかったりで混乱することも予想されます。2番目の疑問についてですが、年限延長は昭和の時代から話題となっていたのですが、諸般の事情でなかなか日の目を見ませんでした。この時期に一挙に6年制が実現したのには種々の理由がありますが、一つは諸外国の薬剤師教育が5年ないし6年が主流になっていることです。米国・フランスでは6年、イギリスでは学部4年で卒業し1年間の登録前研修を行った後、試験を受けて薬剤師となるなど専門職としてのインターン実習が厳しく義務付けられています。我が国の薬学4年制教育の現状は、1ヶ月の実務実習が精一杯で日本の薬剤師免許が世界で通用し難くなっているという現実があります。(ある国で3年で薬剤師免許取得できるとして、その人が日本でも薬剤師と認めて欲しいと主張しても?なのと同じでしょうか。)さらに、医薬分業(平成5年には16%でしたが平成15年には50%を超えた。)により薬剤師の職務が高度かつ複雑になったこと、「薬害」といわれる事例が次々に発生し資質の高い薬学専門家が必要とされていること、医療法や薬剤師法が改正され情報提供、インフォームド・コンセントの義務・努力が課せられたこと、大学・医療現場・文科省・厚労省などの足並みが揃ったことなどが要因と思われます。しかし、実務実習のための教員・場所は確保できるのか、6年制となって学生に来てもらえるのか・学生の負担が増えるのではないか、社会に薬剤師の需要はあるのか、薬剤師免許なしの4年制コースは他学部と差別化できるのか、などの問題をかかえたままのスタートであることも事実といえます。平成16年2月の中央教育審議会における「薬学教育の改善・充実について(答申)」に今回の制度改革の骨格が示されていますが、その中で注目すべき点に「実務実習6ヶ月・共用試験・第三者評価」があります。実務実習6ヶ月実施の中身は、1ヶ月の事前実習・2.5ヶ月の病院実習・2.5ヶ月の薬局実習であり「指導者(教員・薬剤師)・場所・経費」の全てについて現在関係する全ての組織が全力で現在もその準備を行っています。
CBT(Computer Based Testing) OSCE(Objective Structured Clinical Examination) が共用試験ですが、4年次の後期を中心としてそれぞれ1日をかけて実施される予定です。ただし、全国同一日程で行うものではなく、またこの試験で何点以上取らなければ実務実習は絶対受けられないといった評価法の導入には疑問も出ており今後の課題を多く抱えているのが実情です。教養教育が十分か、専門教育は適切か、実務実習はきちんと行われているか、社会から信頼されるにたる大学であることが広く第三者により評価されることにもなっております。 |
回答:国際委員会 |
|
|
|
|
8.20 製剤や機械に関するいろいろな情報を得たいと思います。どんなジャーナルがあるのか、代表的なものを教えてもらえませんか |
|
A: |
例えば、製剤については日本薬剤学会や日本薬学会の会誌「薬剤学」や「ファルマシア」などがあり、製剤と機械の両方では「PHARM TECH JAPAN」((株)じほう)や本会の会誌「製剤機械技術研究会誌」などがあります。また、「粉体と工業」(粉体と工業社)や「粉体工学会誌」(粉体工学会)などのように、必ずしも医薬・製剤に限定したものではありませんが、粉粒体全般を取り扱う機械についての雑誌・会誌があります。その他、医薬品産業界に向けた医薬マーケティング誌「ミクス」(エルゼビア・ジャパン(株))といった雑誌もあります。以下に代表的な雑誌をご紹介します。
| 団 体 名 |
雑 誌 名 |
| (社)日本薬剤学会 |
・薬剤学
・Journal of Drug Delivery Science and Technology (JDDST) |
| (社)日本薬学会 |
・Chemical & Pharmaceutical Bulletin
・Biological & Pharmaceutical Bulletin
・YAKUGAKU ZASSHI
・ファルマシア |
| 日本DDS学会 |
・Drug Delivery System
・Journal of Controlled Release (JCR) |
| (社)日本薬理学会 |
Journal of Pharmacological Sciences |
| 製剤機械技術研究会 |
製剤機械技術研究会誌 |
| (株)じほう |
PHARM TECH JAPAN |
| 粉体工学会 |
・粉体工学会誌
・Advanced Powder Technology |
| エルゼビア・ジャパン(株) |
ミクス |
| (株)粉体と工業社 |
粉体と工業 |
| (社)化学工学会 |
・化学工学
・化学工学論文集
・Journal of Chemical Engineering of Japan |
| (株)工業調査会 |
化学装置 |
| (株)技術情報協会 |
PHARMSTAGE |
| American Pharmacists Association |
Journal of Pharmaceutical Sciences |
| PharmTech |
Pharmaceutical Technology |
| International Society for Pharmaceutical Engineering |
Pharmaceutical Engineering |
Elsevier
(エルゼビア・ジャパン本社) |
・European Journal of Pharmaceutical Sciences
・International Journal of Pharmaceutics
・Powder Technology
・Chemical Engineering Science |
| Taylor & Francis |
Drug Development and Industrial Pharmacy |
|
回答:国際委員会 |
|
|
|
|
8.19 ゲノム創薬という言葉を聞きますが、通常の新薬開発とどこが違うのでしょうか |
|
A: |
従来の創薬手法は、疾病の病態研究から受容体拮抗や酵素阻害活性を指標に数万種類の化合物をスクリーニングする偶然性に頼っていました。近年の分子生物学や情報科学(バイオインフォマティクス)の進展によりゲノム(遺伝子)の解読が進み、癌や糖尿病、高血圧など数多くの疾患に遺伝子が関連していることが明らかになってきました。これらのゲノム情報を活用することにより、疾病関連遺伝子及びタンパク質を特定し、医薬品の標的が明確になり、論理的・効率的に創生することをゲノム創薬といいます。更に病気の原因や薬物応答の個人差の原因となる遺伝子をみつけ、より効果が高く、副作用の少ない医薬品の創生が可能となるゲノム創薬によるテーラーメード医療が期待されます。 |
回答:国際委員会 |
|
|
|
|
8.18 方が改正されましたが、どのような手続きや手順を経て改正されるのでしょうか |
|
A: |
日本薬局方は、薬事法(昭和35年法律第145号)第41条第1項の規定に基づき「医薬品の性状及び品質の適正を図るため」厚生労働大臣が薬事・食品衛生審議会の意見を聴いて定めることになっています。
平成18年3月には(平成18年厚生労働省告示第285号)により第15改正日本薬局方が定められました。
現在は、15局第一追補が19年9月末告示、15局第二追補が20年9月末原案報告のそれぞれ予定で作業が進められています。さらに、23年4月には第16改正日本薬局方施行を目標に準備が始められており、その審議を進めていく上での基本方針が、平成18年8月3日に「第十六改正日本薬局方作成基本方針」として厚生労働省医薬食品局審査管理課から出されています。 ( http://www.mhlw.go.jp/topics/bukyoku/iyaku/yakkyoku/index.html)
この中には、
- 日本薬局方の役割と性格
- 作成方針—日本薬局方改正の5本の柱—
(1)保健医療上重要な医薬品の全面的収載
(2)最新の学問・技術の積極的導入による質的向上
(3)国際化の推進
(4)必要に応じた速やかな部分改正及び行政によるその円滑な運用
(5)日本薬局方改正過程における透明性の確保及び日本薬局方の普及
- 作成方針に沿った第16改正に向けての具体的な方策
等が示されています。
この基本方針に基づき、日本薬局方の原案を作成する際の、具体的な作成方法、記載方法など、第16改正日本薬局方の作成にあたって必要な事項が「第16改正日本薬局方原案作成要領」としてとりまとめられました。( 日本薬局方フォーラム 16巻 別冊 2007 としても発行済 ) ( http://www.pmda.go.jp/operations/notice/2007/jimu20070309.html)
実際の日本薬局方作成は、「厚生労働省、薬事・食品衛生審議会・日本薬局方部会」と「医薬品医療機器総合機構、日本薬局方原案審議委員会」の協力で行われます。更に具体的には、日本薬局方原案審議委員会のなかに、13の委員会が置かれ細部について検討されます。13の委員会名は、化学薬品、抗生物質、生物薬品、生薬等(A)、生薬等(B)、医薬品添加物、理化学試験法、製剤、物性試験法、生物試験法、医薬品名称、国際調和検討、製薬用水、日局標準品です。審議の過程では多くの意見募集も行われています。
公開されている情報をホームページなどで見るとその複雑さに驚いてしまうかもしれません。 |
回答:国際委員会 |
|
|
|
|
8.17 後発医薬品は、先発品と品質が違うという声を聞きます。本来先発医薬品と同等であることが確かめられた上で承認されているはずなの、なぜそのようなことが起こるのでしょうか |
|
A: |
後発医薬品は、人に投与された時に先発医薬品と同等の血中濃度が得られること(つまり同等の効果が期待できる)を生物学的同等試験で確認しております。しかし、通常後発医薬品の製剤処方は、先発医薬品と異なっています。そのため、ある条件下では先発医薬品と溶出性などの特性が異なることが報告されています。生物学的同等性試験は、一定の均一な背景を持った被験者により行われますが、承認され市場に出た場合には、胃酸度が異なる患者さんを含めいろいろな背景を持った患者さんに投与されます。そのため、一部の後発医薬品において製剤処方の違いなどに起因して、同じ効果が得られない場合のあることが明らかとなっています。 |
回答:国際委員会 |
|
|
|
|
8.16 後発医薬品は先発医薬品と同じと聞きますが、適応症が違う場合があります。どうしてでしょうか |
|
A: |
この質問に回答するためには、新医薬品の「再審査制度」について理解する必要があります。「再審査制度」は、製薬企業が、新医薬品が承認された日から通常6年間(オーファンドラッグは10年、新効能・効果・用量・用法は4年)、使用成績などを調査し、その結果に基づき安全性や有効性などについて厚生労働省の方で再度確認する制度です。この期間は、後発医薬品の承認・販売が出来ません。こうした背景があることを念頭において、質問に回答します。
適応症が先発品と異なる場合、2つのケースが考えられます。
- 先発品が途中から効能効果の拡大を行い、この結果既に取られている適用の再審査が終了した後に、追加の適用に対する再審査期間があり、この間後発医薬品は、先発品の追加の効能効果に対する適用が得られないことになります。また、そうした追加の効能効果に対する市場性が小さい場合、仮に追加の効能効果の再審査が終了したとしても、適用に関する一変申請を行わないことがあります。
- 後発医薬品同士で適応症が異なる場合もありますが、これは(1)との関係もあり、追加の効能効果の再審査後に申請した後と前で、つまり後発医薬品の申請時期により異なることがあります。
厚生労働省は、こうした事態について後発医薬品使用の促進から、平成18年3月医政局長通知「効能効果、用法用量等を標準先発品と合致させること」を出しており、今後適応症が異なる場合は少なくなると思われます。
なお、医薬工業協議会が制作している「ジェネリック医薬品Q&A」(監修:厚生労働省)(2006年12月発行)の中では、次のように記載されています。
ジェネリック医薬品と先発医薬品の効能・効果(適応症)等が一致していない場合があります。その主な理由は、先発医薬品の効能・効果の一部に再審査期間や用途特許が付されているため、その効能・効果の承認取得ができないためです。これらの場合を除き、効能・効果の一部が欠けているものは速やかに取得するよう製造販売業者に求められています。 |
回答:国際委員会 |
|
|
|
|
8.15 くすりの数は、非常に多いと思いますが、処方箋薬局には全ての種類が
おいてあるのでしょうか |
|
A: |
処方箋用の医薬品の種類は数千種類といわれ、これを単独の処方箋薬局で全て準備することはコスト的に成り立つものではありません。処方される医薬品は、病院や医師により異なるため、病院と処方箋薬局で保有薬剤の情報を共有化したり、処方箋薬局間でどのような薬剤をどこが保有しているかという情報をネットワーク化しておき、患者の要望に直ちに対応できるような体制がとられています。 |
回答:国際委員会 |
|
|
|
|
8.14 とまって直すプリザSのコマーシャルで、DDSという言葉を聴きますが、普通のくすりとどこが違うのでしょうか |
|
A: |
DDSとは「 Drug Delivery System(薬物送達システム)」の略で、薬物を目的とする作用部位のみへ選択的に送達する(ターゲッティング)、必要量の薬物を適切な時間かつ適切な部位へ送り届ける(コントロールドリリース)などにより、最適な治療効果を得る事を目的とした、新しい薬物投与技術のことを言います ※参考文献 1。いわゆる普通の薬を服用した場合、薬物は胃液による分解や肝臓での代謝などを受けるため、薬物が100%作用部位へ届くことは極めて稀です ※参考文献 2。これに対してDDSの概念を導入した薬では、機能性添加剤(腸溶性ポリマー、徐放性ポリマーなど)を処方に組み込んだり、剤形(多層錠、マイクロカプセルなど)を工夫することによって、薬物の胃での分解を回避したり、血中濃度を長時間治療濃度域に維持させるなどの効果を得ることが出来ます。ちなみにプリザSの場合では、添加剤であるカルボキシビニルポリマーの働きによって薬剤を患部付近に長時間留まらせることで、有効成分が効率よく患部に働くように工夫が施されています ※参考文献 3 。
《参考文献》
- 辻・河島 編、「わかりやすい物理薬剤学 第3版」(廣川書店) p.239-258、(2002)
- 永井他、「CMCの実際 — 製剤研究のデザイン —」(じほう) p.116-119、(2003)
- 大正製薬ホームページ「プリザS製品情報」より
|
回答:国際委員会 |
|
|
|
|
8.13 新薬の開発には非常にお金がかかるといいますが、どの位かかるのでしょうか |
|
A: |
新医薬品の開発にかかる費用は、資料により若干数値が異なっております。
代表的な数値を上げます。
1.厚生労働省「医薬品産業ビジョンの概要」平成14年8月30日:260億〜360億円
2.山田武「医薬品開発における期間と費用」医薬品産業研究所リサーチペーパー・  シリーズ No.8:350億円
3.日本製薬工業協会「てきすとぶっく 製薬産業2005」:500億円(日本の調査)、 約8億ドル(米国の調査)
このほかインターネットなどで調べると100億〜300億円という数値がみうけられます
。
調査方法により、こうした数値は変動するものと思いますが、概略としては上述の
ような費用がかかるものと考えられます。
参考資料:
M.Dickson, J.P.Gagnon, The Cost of New Drug Discovery and Development,
Discovery Medicine, 4(22) 172-179(2004)
Editorial, New estimates of drug development costs, J.Health Economics, 22, 325-330 (2003) |
回答:国際委員会 |
|
|
|
|
8.12 プレフォミュレーションについて、具体的にどのようなことをやるのでしょうか |
|
A: |
プレフォミュレーション(preformulation:前処方化)とは、“至適な投与製剤の設計を行うために原薬の諸特性を明らかにし、得られた知識を確実に製剤設計の面に反映させる過程“(プレフォーミュレーションと薬物物性試験(医薬品開発 16巻)、廣川書店 1990)であるということが出来ます。プレフォーミュレーションで検討される内容としては、1)溶解性や分配係数など薬物の物理化学的な性質、2)薬物の安定性など処方設計に関わる物理化学的な性質、3)薬物の吸収性や消化管内での安定性など薬物の生物薬剤学的特性が含まれます。こうした検討の具体的な方法や内容については、橋田充(編) 経口投与製剤の設計と評価 薬業時報社、p69—p165(1995)に詳細に記載されています。また、「上述の医薬品開発 16巻」にも詳細に記載されていますので、参照してください。 |
回答:国際委員会 |
|
|
|
|
8.11 日本薬局方は、なぜ「法」とせずに「方」の字を用いたのでしょうか |
|
A: |
薬局方の語源につきましては、エーザイ 薬博物館 HPにあります “もうひとつの学芸員室”の中に、次のように記載されています。
薬局方は、薬剤師にとってバイブルのようなもの、必要不可欠な医薬品の情報が記された馴染み深い公定書であり、世界主要各国(あるいは数カ国共同)がそれぞれの薬局方を発行しています。我が国の『日本薬局方』は、薬事法によって医薬品の性状及び品質の適正を図るために、厚生労働大臣が薬事・食品衛生審議会の意見を聴いて定めた医薬品の規格基準書として、通則、製剤総則、一般試験法及び医薬品各条からなり、繁用されている医薬品が収載されています。最新版は、平成13年3月30日に公布された第十四改正ですが、明治19年(1886)6月に初版が公布されて以来、医薬品の開発、試験技術の向上にあわせて内容の改訂がくりかえされてきました。最初の『日本薬局方』は、お雇い外国人の化学者ゲールツらの指導によってドイツ薬局方、オランダ薬局方を参考に作成されました。海外から、不良品、粗悪品などを含むさまざまな品質規格の医薬品が大量に輸入され、品質を統一する基準が必要になったためです。
くすり博物館では、稀少な119年前の初版『日本薬局方』を保存しています。明治19年6月25日の官報第894号別冊内務省令第10号の官報に掲載されたのが最初ですが、[欧米各国局方対照][音釈付][註釈]などを掲載したものや、ラテン語に翻訳された薬局方もあります。こうした『日本薬局方』の発布は、明治憲法発布に先立つ3年前のことで、近代的スタイルのものとしては、東洋では最初、世界では第21番目の国定薬局方でした。そもそも「薬局方」という名称、Apotheek(オランダ語)、Pharmacopoea(ラテン語)、英語では、Pharmacopoeia(英語)、Pharmacopeia(米語)、ですが、語源はギリシャ語の「薬」と「作り方」に由来するといわれています。日本語の「局方」という独特の名称がなぜ、「公定薬典」ではなく「局方」なのか。また、「法」とせずに「方」の字を用いたのかと不思議に思う方もいますが、江戸中期、蘭方医中川淳庵が オランダの薬局方「アポテーキ」を「和蘭局方」と訳したのが、書名での最初の使用といわれています。さらに起源を遡ると、中国宋代(1078-85)に刊行された協定処方集『(太平惠民)和剤局方』に倣ったものだとされています。『和剤局方』は、日本にも平安末期に伝わり、漢方製剤の適応症、薬剤名、処方量、調製法、用法用量などについてが詳述されていて、現在の薬局方のような書物として江戸時代から明治初期に利用されていました。 |
回答:国際委員会 |
|
|
|
|
8.10 開発化合物の品質試験を外部へ委託する場合、GLP準拠とGMP(あるいは治験薬GMP)準拠でどのような違いがあるのでしょうか? また、外部機関への要求内容などの違いについて説明してください |
|
A: |
まず始めにGLPについてですが、開発品の有効性、安全性(毒性)等の確認のための動物試験に対し、遵守すべき事項がこのGLPで規制されています。動物実験だけでなく、薬物の血中濃度の測定(TK試験)、投与する薬物の品質試験及び動物に投与する際の投与液の濃度確認もGLPの対象となります。これらのGLP試験のうち、動物実験とTK試験を実施するためには、施設、設備について当局から認証(総合機構の適合性評価)を受けなければなりません。即ち、認証を受けた施設、設備でなければ、GLP試験は実施できません。ただ、投与する薬物の品質試験及び動物に投与する際の投与液の濃度確認試験までは、認証の施設、設備での試験要求はありませんが、試験に当っては当局からのGLP省令を遵守する必要があります。
次にGMPについてですが、医薬品の製造管理、品質管理に対し、遵守すべき事項がGMPで規制されています。市販薬についてはGMP、開発品については治験薬GMPとして区別されています。昨年の薬事法改正に伴い、市販薬の製造あるいは試験施設については、当局からGMPGMP適合性調査を受け、登録されることになっています。ただし、製造業の業許可更新まではみなし対応がなされていますが、業許可更新時にGMP適合性調査を受けることになります。治験薬GMPについては、このGMP適合性調査はありませんので製造あるいは試験施設の登録の必要性はないようですが、治験薬GMPの規則内容は遵守しなければなりません。
外部機関への要求内容については、試験に関する逸脱や異常報告などの連絡体制や、取り決めをしておき、定期的な監査システムなどを盛り込んだ契約を締結しておくべきでしょう。 |
回答:国際委員会 |
|
|
|
|
8.9 Regulatory Science とは、何をさすのでしょうか |
|
A: |
レギユラトリーサイエンスは,科学技術の所産を人間の生活に取り入れる際に,最も望ましい形に調整するための科学である。
財団法人日本薬剤師研修センター 内山充理事長は、「レギュラトリーサイエンスの発展—官・学・産のフォーラムを目指して—」(エルゼビア・ジャパン株式会社発行)の中で下記のように述べています。
医薬品や食品に関する厚生行政の施策はすべて科学を根拠とし、そこで行われる規制や許認可は、安全で有用なものを正しく評価して優れた品物を速やかに国民に提供するためのもの、すなわち、科学技術を国民生活に調和させ、安全に利用するための作業である。レギユラトリーサイエンスはその根拠を明らかにするための科学であるので「行政を支援するための試験研究」と理解することもできる。
このような意味での研究を適切に行うためには、広い視野と最新の知識が必要であり、それを支える研究活動が常時行われていなければならない。それらの研究では、評価に必要な技術や指標の確立のほか、材料、物質等の動向を予測し、その特質を考慮した評価研究が必要であり、既存の基礎科学にも応用科学にもない独自の研究分野を形成するものである。すなわち,レギユラトリーサイエンスの研究内容は「評価科学」であり、実践の場では「行政科学」と言い換えることもできる。
厚生省の試験研究機関として当所(国立医薬品食品衛生研究所)は、医薬品や医療用具、食品や環境中に存在する各種化学物質など、人間の生命・健康に直接影響を及ぼすものを研究対象とし、大学や企業の研究機関では研究対象とし難い分野を含め、多くの基礎研究からアセスメントに至る広い範囲を照準に入れたレギユラトリーサイエンスを業務としている。
レギユラトリーサイエンスにおいては、標準的試験法や判定法の開発は重要な一分野であるが、そのためには種々の物質の反応や、生体あるいは病態での機能解析ないしは機構解明に関する先導的,基盤的な研究が行われていなければならない。そしてそれらはガイドラインや公的基準に結びつく適切な予測理論と、それを支える正確で豊富な物理的、化学的、あるいは生物学的実験根拠が付随している必要がある。さらに、それらは学問的な評価に耐えうるものでなければならない。
一方、レギユラトリーサイエンスの基礎研究に基づいて行われるレギユレーションは、今日もはや我が国一国だけの問題としては通用しない。国際的に矛盾のない妥当な結論が要求される。そこでは前例や形式は根拠とならない。あくまでも科学的根拠のみが共通の言葉として通用する。国際会議における討論は、レギユラトリーサイエンスに携わるものにとって極めて有益なフイードパック情報である。
多くの問題においてレギユラトリーサイエンスでの評価手法が求められている。レギユラトリーサイエンスの目標とするところが正しく実践されて、国民生活の向上に反映されると同時に、我が国の科学技術の発展に寄与することができれば幸いである。 |
回答:国際委員会 |
|
|
|
|
8.8 米国における健康保険システムは、日本とはずいぶん異なると聞きます。どう違うのか教えてください |
|
A: |
日本は、政府管掌保険や組合健康保険を中心とした皆保険制度を採用しておりますが、米国では民間任意保険(競争原理に基づく保険)が中心となっています。その為、収入の低い高齢者や発病率の高い人は高い保険料が払えないケースが出て1960年代に社会問題となり、1965年政府管掌のメデイケア(Medicare)(65歳以上の高齢者)とメディケイド(Medicaid)(貧困者・障害者)という保険が発足しました。 しかしながら、現在でも米国では20%近くの人が保険に入っていないといわれ、国民皆保険制度の日本とはまったく異質のものとなっています。 医療費抑制が叫ばれる中、米国における民間保険では、HMO(Health Maintenance Organization)やPPO(Preferred Provider Organization)が主流になり、患者の自由な診療等が制限される状況も生まれ問題となっています。
(参)
渡辺敏一、アメリカ医療保険物語(5回シリーズ)、月刊薬事、39(6)-(11)、(1997) |
回答:国際委員会 |
|
|
|
|
8.7 In vivoでの同等性試験は、なぜ動物ではダメなのでしょうか |
|
A: |
ここでは、経口投与製剤を念頭において回答致します。生物学的同等性試験において、ヒトの代わりとなる動物が備えるべき条件は、
(1) 患者に投与する製剤を服用できる
(2) 製剤間のバイオアベイラビリティの差がヒトにおける差よりも大きい
(3) 検出力が高い
などでしょう。従って、ウサギ以上の大きさの動物となりますが、実際的には、ウサギ、ミニブタ、イヌ、サルが考慮の対象となります。ウサギは糞を食べるという習慣があるために、胃内容排出速度がヒトと全く異なり、ウサギの実験からヒトにおける製剤の生物学的同等性を予測することはできません。イヌについては、ヒトに比べて、吸収部位が短い、食事時間との関係で胃内容排出時間が異なる、消化管内における製剤に対する破壊力が強いなどの報告がなされています。これらは、すべて製剤間のバイオアベイラビリティの差を小さくする方向に作用し、ヒトの生物学的同等性を予測するためのスクリーニング動物として、(2)の条件を満たさず不適切です。ミニブタ、サルでは、ヒトによる試験結果と一致するというデータが十分に揃っていません。以上のような理由から、ガイドラインでは、生物学的同等性試験に動物を使用することは認めておりません。しかし、ガイドラインはこのような限界を知った上で、医薬品の開発段階で探索的に動物を使用し、バイオアベイラビリティの検討を行うことを妨げるものではありません |
回答:国際委員会 |
|
|
|
|
8.6 CROというのは何ですか?その役割等(どんな所で活用したらよいか)について教えてください |
|
A: |
製薬企業などから医薬品などの開発業務を受託する「開発業務受託機関(企業)」をCROと呼びます。英語でContract Research Organization と表し、その頭文字をとったものです。
医薬品、医療用具、化粧品などの臨床試験や市販後調査を実施する医療機関に関わる様々な業務を受託しています。
日本CRO協会のホームページ( http://www.jcroa.gr.jp/)に掲載されている「紹介パンフレット」が参考になります。 |
回答:国際委員会 |
|
|
|
|
8.5 米国における特許制度について教えてください |
|
A: |
米国特許制度の特徴
(1)先発明主義
同一発明について二以上の特許出願人がいたとき、最も先に発明した者(発明者)のみが特許を受けられる。尚、先発明主義を廃止し、先願主義に移行する法案が米国下院に提出されており、今後の動向が注目される。
(2)特許出願人
特許出願は発明者がしなければならない。発明者が属する企業の名義で特許を取得するためには、特許出願をした発明者から特許を受ける権利を企業に譲渡しなければならない。
(3)情報開示義務
特許出願に関与した者は、自己の知っている先行技術を米国特許商標庁に提出しなければならない義務を負っている。
(4)審査請求制度はない。仮出願制度、継続審査請求制度がある。
(5)判例法主義
米国では「法は根源的に判例から発生する」という判例法主義がとられている。従って、新たな判例により法律の解釈が変動していく。
(6)陪審員制度
特許侵害訴訟における当事者は陪審審理を請求することができる。
(7)故意侵害
裁判所は、故意に特許権を侵害した者に対して損害賠償額を3倍まで引き上げることができる。
2.米国特許取得までの手続の流れ(下記図参照)
適式な米国特許出願を提出することによって、米国特許出願日が付与される。米国にした仮出願、外国(パリ条約加盟国)にした特許出願に基づく優先権を主張して米国特許出願をすることができる(12ヶ月以内)。米国特許出願は、原則として、優先日から18ヶ月経過後に出願公開される。
日本や欧州のような審査請求制度はなく、米国特許出願が提出されると自動的に審査に係属する。審査官は、まず、特許出願のクレームが発明の単一性を有するか否かを審査し、一出願に複数の発明が含まれていると判断した場合は、限定要求が出願人に通知される。この限定要求に対して、出願人は審査対象とするクレームを選択しなければならない。
審査官は、つぎに、クレームに記載された発明が法定の特許要件(記述要件、新規性、非自明性等)を備えているか否かを審査する。審査の結果、特許要件を備えていないと判断した場合は、最初の局指令(拒絶理由通知)が出願人に通知される。これに対して、出願人はクレームの補正や意見書の提出などによって反論することができる。この反論によっても拒絶が解消しない場合、最終局指令が出願人に通知され、この最終局指令に対する反論によってもなお拒絶が解消しない場合は、アドバイザリーアクションが出願人に通知される。アドバイザリーアクションに対して、出願人は審判(拒絶査定不服審判)を請求するか、継続審査請求を請求することができる。継続審査請求を請求した場合、最終拒絶の状態は解消され、再度審査官による審査に付される。審判の審決に対しては、審決取消訴訟で争うことができる。
一方、特許が許可可能であると審査官が判断した場合は、特許許可通知が出願人に通知される。そして、出願人が特許発行料の支払いをすると、特許が発行され、特許公報に掲載される。 |
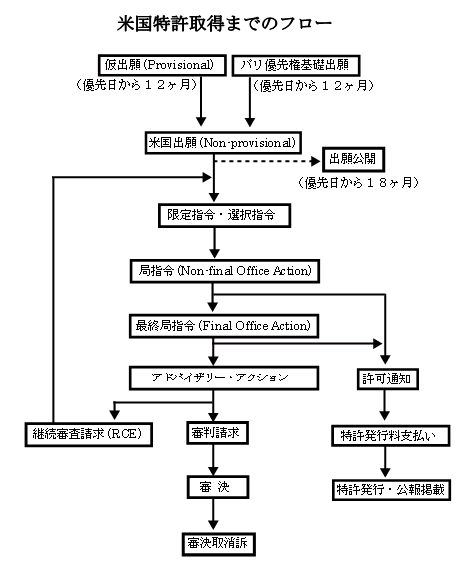 |
回答:国際委員会 |
|
|
|
|
8.4 東南アジアの行政当局の情報はどこで入手できますか。解説もあまり出ていないようですが? |
|
A: |
確かに東南アジアの薬事行政情報については纏まった解説など少なく、どこに問い合わせたらいいかわからない方が多いと思います。東南アジアの最新の行政情報は日本製薬工業協会(製薬協)のアジア部会に問い合わせて見て下さい。最新の情報が得られると思います。
東南アジアには日本製薬企業の進出も多く、現地の日系企業は日本の親会社と緊密な連絡を取っており、その情報は製薬協のアジア部会に集まっています。
また、2002年9月30日(株)情報機構から発刊された下記の専門書も参考になると思います。
「アジアにおける医薬品事業の展開」
—医療・薬事の最新事情から事業戦略の指針まで—
(ISBN 4-901677-03-9)
出版元:情報機構
〒141-0032 東京都品川区大崎5-4-18
朝日プラスチック東京本社ビル4F
TEL:03-5740-8755/FAX:03-5740-8766
Eメール:req@johokiko.co.jp |
|
回答:--- |
|
|
|
|
8.3 いろいろな用語、例えばGMP、QA、OQ・・・・、が日常茶飯事に使われますが、学会等で公に認められている日本語訳はあるのでしょうか。製機研のホームページにもあるようですが? |
|
A: |
機研、PDAやISPE等いろいろな団体が、それぞれの資料・情報に基づいて訳を行っておりますが、一般には厚生労働省等の政府が発行する法律・指針等の中で使用される訳が公に認められたものと考えることが出来ます。 |
回答:--- |
|
|
|
|
8.2 米国には薬価がないと聞きますが、薬の値段はどのようにして決められるのでしょうか |
|
A: |
米国には、日本の薬価のような公定価格というものはありません。保険組合等の問題を含め、薬の価格を簡単に述べることは難しいのですが、一般的に薬の値段は、製品を販売する会社が、その製品のコスト・有用性、先発品の価格、販売戦略などに基づいて設定します。従いまして、患者が求める有用性の高いものであれば、高額な設定も可能です。一方、ゾロ品に近い場合、市場における浸透性を図るような戦略から低価格にするケースもあります。例えば、レスコール(ノバルティスファーマ)('94.4)の上市価格は$1.02ですが、これは主要競合品(メバコール:$1.98)と比較し、49%も低いものでした。また、プレバシット(タケダ・アボット)('95.6)($3.25)は、プリロゼック(アストラゼネカ)($3.63)よりも10%低いものでした。逆に、フルナーゼ(グラクソ)(1.95)($38.88)は、パンセナーゼ(シェリング)($32.40)よりも20%高い価格が設定されました。
(参)E. M. Kolassa著 佐賀見國一監修「米国における医療用医薬品価格戦略—日米の比較も含めて—」じほう(2001) |
回答:--- |
|
|
|
|
8.1 ICHに当製剤機械技術研究会から代表を送ることは可能でしょうか |
|
A: |
不可能ではありませんが、正式なメンバーは確定されておりますので、こうしたメンバーからの要請を受けての派遣となると思います。過去に、ISPEのメンバーが参加した例があります。 |
回答:--- |
|
|
|
