| 国 | 日本 | 日本 | 日本 | 米国 | ― | 欧州 | ― | ― | ― | ― | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | EUROPEAN COMMISSION | WHO | WHO | WHO | WHO | ||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品 の製造に関する指針 |
最終滅菌法による無菌医薬品 の製造に関する指針 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice
Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 44th report Annex 3 WHO good manufacturing practices for pharmaceutical products containing hazardous substances WHO Technical Report Series, No. 957, 2010 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
||||||||||||||
| 発行年 | 2011年 | 2012年 | 2025年 | 2004年 | 2023年 | 2022年 | 2010年 | 2018年 | 2022年 | 2019年 | ||||||||||||||
| 記載内容 | 6.構造設備 6.1 構造設備の設計上の要点 26) 更衣室はエアロックの機能を設け、(中略)更衣に伴う一次的な微粒子の増加を早く低減させるため、更衣室の空間体積や換気回数(回復時間)を考慮するとともに更衣室内の空気は上部から供給し下部から排気することが望ましい。 |
7.無菌医薬品に係る製品の作業所 7.2 空調システム 7.2.2 空気 7) 無菌操作区域の製造作業室及び更衣室においては、作業内容の製品に対する汚染リスクを評価し、定められた清浄度レベルを維持するために適切な換気回数を設定すること。 通常、直接支援区域では30回/時間、その他の支援区域の内、グレードCに相当する作業室では20回/時間を確保することが望ましい。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 25) 更衣室は、エアロックの機能を設け、(中略)更衣に伴う一次的な微粒子の増加を早く低減させるため、更衣室の空間体積や換気回数(回復時間)を考慮するとともに更衣室内の空気は、上部から供給し下部から排気することが望ましい。 |
4 建物 4.14 クリーンルームは、(略) その給気は当該区域を効果的に換気するものであること。 |
クリーンルーム及び清浄空気設備の適格性評価 4.25 クリーンルーム及び清浄空気設備の適格性評価は、等級分けされたクリーンルーム又は清浄空気設備がその用途に適合するレベルを評価するプロセス全体である。アネックス15の適格性評価要件の一部として、クリーンルーム及び清浄空気設備の適格性評価には(当該設備の設計/運用に関連する場合において)以下を含めること: (略) viii. 回復試験 (略) |
4.32 クリーンルーム及び清浄空気設備の適格性再評価が、所定の手順に従って定期的に行われていること。その適格性再評価には最低限、以下を含めること: (略) v. 空気速度試験 (注:グレードB、C及びDについては、CCSの一環として文書化されたリスク評価に従って、空気速度試験が行われていること。ただし、一方向気流が供給される容器充填区画(例:最終滅菌法による製品を容器充填する際、又はグレードA及びRABSのバックグラウンド)には、空気速度試験が要求される。 気流が一方向でない等級については、回復試験の計測で速度試験を代替すること。 (略) |
IV. Buildings and Facilities C. Clean Area Separation (略) Air change rate is another important cleanroom design parameter. For Class 100,000 (ISO 8) supporting rooms, airflow sufficient to achieve at least 20 air changes per hour is typically acceptable. Significantly higher air change rates are normally needed for Class 10,000 and Class 100 areas. |
4 Premises 4.14 Cleanrooms (略) should flush the area effectively. |
CLEANROOM AND CLEAN AIR EQUIPMENT QUALIFICATION 4.25 Cleanroom and clean air equipment qualification is the overall process of assessing the level of compliance of a classified cleanroom or clean air equipment with its intended use. As part of the qualification requirements of Annex 15, the qualification of cleanrooms and clean air equipment should include (where relevant to the design/operation of the installation): (略) viii. recovery test, |
4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include at a minimum the following: (略) v. air velocity test (Note: For grade B, C and D the air velocity test should be performed according to a risk assessment documented as part of the CCS. However, it is required for filling zones supplied with unidirectional airflow (e.g. when filling terminally sterilised products or background to grade A and RABS). For grades with non-unidirectional airflow, a measurement of recovery testing should replace velocity testing). (略) should flush the area effectively. |
4 Premises 4.14 Cleanrooms (略) should flush the area effectively. |
Cleanroom and clean air equipment qualification 4.25 Cleanroom and clean air equipment qualification is the overall process of assessing the level of compliance of a classified cleanroom or clean air equipment with its intended use. As part of the qualification requirements of Annex 15, the qualification of cleanrooms and clean air equipment should include (where relevant to the design/operation of the installation): (略) viii. Recovery test. |
4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include at a minimum the following: (略) - Air velocity test (Note: For grade B, C and D the air velocity test should be performed according to a risk assessment documented as part of the CCS. However, it is required for filling zones supplied with unidirectional airflow (e.g. when filling terminally sterilised products or background to grade A and RABS). For grades with non-unidirectional airflow, a measurement of recovery testing should replace velocity testing). |
9. Air-handling systems 9.7 There should be a system description including schematic drawings detailing the filters and their specifications, the number of air changes per hour, pressure gradients, clean room classes and related specifications. These should be available for inspection. |
4. Premises 4.6 HVAC systems should ensure that the specified room conditions are attained, for example through heating, cooling, air filtration, air distribution, airflow rates and air exchange rates. |
5. Design of HVAC systems and components 5.16 Periodic switching off of AHUs, for example, overnight or at weekends, or reducing supply air volumes during non-production hours, should be avoided so that material or product quality is not compromised. Where AHUs are switched off, there should be appropriate justification and no risk to materials or products. The procedure and its acceptability should be proven and documented. |
12. Qualification 12.8 There should be standard operating procedures describing the action to be taken when alert and action limits are reached. This may include, where relevant: (略) ・room air-change rates; |
4. Premises 4.14 Cleanrooms (略) should flush the area effectively. |
Cleanroom and clean air equipment qualification 4.25 Cleanroom and clean air equipment qualification is the overall process of confirming the level of compliance of a classified cleanroom or clean air equipment. As part of the qualification requirements, the qualification of cleanrooms and clean air equipment should include (where relevant to the design and operation of the installation): (略) viii. recovery test |
4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include, at a minimum, the following: (略) v. air velocity test. Note: For grade B, C and D, the air velocity test should be performed according to a risk assessment documented as part of the CCS. It is however, required for filling zones supplied with unidirectional airflow (for example, when filling terminally sterilized products or background to grade A and RABS). For grades with non-unidirectional airflow, a recovery test should replace velocity testing. |
4. Premises 4.2 Weighing/dispensing and sampling areas (略) To further support containment, consideration may also be given to having material airlocks (MALs) and personnel airlocks (PALs), where needed, for entry and exit of processing areas (for an example, see Fig. A2.6). Appropriately designed airlocks can assist in ensuring containment. Additional controls, such as pressure differentials between areas, an appropriate number of air changes in an area, and sufficient filtration of air, should be in place. |
(続き、略) Washing areas should be designed and used in such a manner that equipment and components will not be re-contaminated after cleaning. The system supplying and extracting air from the area(s) should be suitably designed to ensure that this objective is achieved. Principles that may be considered include (but are not limited to) filtration of air, pressure differentials between areas, air changes per hour and airflow directions (for an example, see Fig. A2.7). |
7. Air filtration, airflow direction and pressure differentials (略) The number of air changes or air-exchange rates should be sufficient. A guidance value is between 6 and 20 air changes per hour. Manufacturers should also establish how much time it takes for a room that is out of its classification to return within the specified class. This is often referred to as clean-up or recovery time. A guidance time period for clean-up or recovery is about 15-20 minutes. |
12. Qualification (略) Some of the typical HVAC system parameters that should be included in the tests during qualification are listed next and the selection of the parameters should be justified (for examples, see Table A2.3). It is recommended that the tests be done at defined intervals. The tests typically cover: (略) ・room air-change rates; |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・ 更衣室の空間体積や換気回数(回復時間)および上部給気・下部排気。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌操作区域の製造作業室及び更衣室において通常確保すべき換気回数。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・ 更衣室の空間体積や換気回数(回復時間)および上部給気・下部排気。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの給気による効果的な換気。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・回復試験の適格性評価。 *回復試験に関連する「換気回数」の具体的数値表記はありません。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・回復試験の適格性再評価。 *回復試験に関連する「換気回数」の具体的数値表記はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・清浄度区分における最低換気回数。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.25 viii. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32 v. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.25 viii. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32 v. と同じ内容となります。 |
対象医薬品:有害物質含有医薬品 以下の要件があります。 ・換気回数を含む空調システム仕様の記述が必要であり、査察時に利用できること。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調システムにおいて設定されたとおりの換気回数などが確実に達成さること。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・夜間や週末における空調機の定時停止や非生産時の給気風量の低減運転および空調を止める場合の条件について、原料・製品へのリスクがないようにすること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・換気回数その他において、アラートレベル、アクションレベルに達した際にとるべきアクションについて、SOPに記述すること。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.25 viii. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32 v. と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・秤量/小分けおよびサンプリング区域における封じ込めにおけるエアロックの換気回数。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・洗浄室エリアの空調について清浄化済みの装置や部品が再汚染しないよう換気回数を設定すること。 *「換気回数」の具体的数値表記はありません。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・換気回数のガイダンス値(6〜20回/h)、回復時間(リカバリータイム)のガイダンス値(15〜20分)。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・換気回数は、空調システムのパラメータのうち、適格性評価の検査対象となる。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | 米国 | ― | ― | ― | ― | ― | 欧州 | 欧州 | 欧州 | ― | ― | ― | ― | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CDER | PIC/S | PIC/S | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | WHO | WHO | ||||||||||||||||||||||||||||||||||||||||
| 分 類 | 省令 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | Code of Federal Regulations | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
医薬品及び医薬部外品の 製造管理及び品質管理の 基準に関する省令 |
薬局等構造設備規則 | 無菌操作法による無菌医薬品 の製造に関する指針 |
最終滅菌法による無菌医薬品 の製造に関する指針 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2A ヒト用先端医療医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2B ヒト用生物学的医薬品( 原薬及び製剤) の製造 |
PIC/S のGMPガイドラインを活用する際の考え方について 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS
ANNEX 2A MANUFACTURE OF ADVANCED THERAPY MEDICINAL PRODUCTS FOR HUMAN USE |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS
ANNEX 2B MANUFACTURE OF BIOLOGICAL MEDICINAL SUBSTANCES AND PRODUCTS FOR HUMAN USE |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS
Annex 3 MANUFACTURE OF RADIOPHARMACEUTICALS |
EU Guidelines to Good Manufacturing Practice
Medicinal Products for Human and Veterinary Use Part 1 |
EU Guidelines to Good Manufacturing Practice
Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 3 Manufacture of Radiopharmaceuticals |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 54th report Annex 2 International Atomic Energy Agency and World Health Organization guideline on good manufacturing practices for radiopharmaceutical products WHO Technical Report Series, No. 1025, 2020 |
||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2021年 | 2021年 | 2011年 | 2012年 | 2020年 | 2025年 | 2022年 | 2022年 | 2012年 | 2023年 | 2004年 | 2021年 | 2023年 | 2023年 | 2023年 | 2023年 | 2014年 | 2022年 | 2008年 | 2022年 | 2018年 | 2019年 | 2020年 | ||||||||||||||||||||||||||||||||||||||||
| 記載内容 | 第三章 医薬部外品製造業者等の製造所における製造管理及び品質管理 第三節 無菌医薬部外品の製造管理及び品質管理 (令三厚労令九〇・追加) (無菌医薬部外品の製造所の構造設備) 第五十一条 三 作業室は次に定めるところに適合するものであること。 ハ 無菌操作を行う区域は、フィルターにより処理された清浄な空気を供し、かつ、適切な差圧管理を行うために必要な構造設備を有すること。 |
第二章 医薬品等の製造業 第一節 医薬品の製造業 (特定生物由来医薬品等の医薬品製造業者等の製造所の構造設備) 第八条 一 特定生物由来医薬品等に係る製品の製造所(包装、表示又は保管のみを行う製造所を除く。)は、次に定めるところに適合するものであること。 ホ 作業所のうち、無菌操作を行う区域は、フイルターにより処理された清浄な空気を供し、かつ、適切な差圧管理を行うために必要な構造及び設備を有すること。 ヘ 作業所のうち、病原性を持つ微生物等を取り扱う区域は、適切な陰圧管理を行うために必要な構造及び設備を有すること。 |
第四節 再生医療等製品の製造業 (再生医療等製品製造業者等の製造所の構造設備) 第十四条 十一 作業所のうち、無菌操作を行う区域は、フイルターにより処理された清浄な空気を供し、かつ、適切な差圧管理を行うために必要な構造及び設備を有すること。 十二 作業所のうち、病原性を持つ微生物等を取り扱う区域は、適切な陰圧管理を行うために必要な構造及び設備を有すること。 |
6.構造設備 6.1 構造設備の設計上の要点 19) 清浄区域にはそこで行われる作業に対して適切な清浄度レベルを維持するためHEPAフィルター等の適切なフィルターによりろ過した空気を供給し,適切な室間差圧を設けること.また,室間差圧が維持されていることを監視できるようにすること. 22) 清浄区域の室圧は扉などで隣接する清浄度レベルの低い区域の室圧よりも高く設定すること.ただし,封じ込め施設の場合はこの限りではない. |
7.無菌医薬品に係る製品の作業所 7.2 空調システム 7.2.2 空気 各清浄区域の環境を維持するためには,清浄度レベルの高い区域から,隣接する清浄度レベルの低い区域へ流れる適切な気流を確保することが重要である. 1) 無菌操作区域とその他の支援区域との間の室間差圧を設定し,管理及び監視を行うこと. 2) 無菌操作区域とその他の支援区域との間にはエアロックを設け,室間差圧及び気流の逆転が起きないよう,十分な差圧を設けること.扉を閉じた状態で10〜15Pa又はそれ以上の差圧を維持することが望ましい.当該エアロックの設計は,6.1 26)更衣室に準じる. その他の支援区域内においても空気の清浄度レベルの異なる区域の間には,適切な差圧を設けること. |
(続き) 3) 製品の無菌性を確保する上で特に重要と考えられる差圧については,モニタリングを常時行うこと.さらに,異常時に備えて警報システムを備えることが望ましい. 9) 製造作業中の差圧変動及び気流パターンを定めて文書化し、実際の差圧及び気流の状態が工程に適したものであることを実証すること。また、職員の介入による乱気流が環境の清浄度レベルに及ぼす影響について検討し、当該作業に係る手順書に反映すること。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 18) 清浄区域には、そこで行われる作業に対して適切な清浄度レベルを維持するためHEPAフィルター等の適切なフィルターによりろ過した空気を供給し,適切な室間差圧を設けること.また室間差圧が維持されていることを監視できるようにすること. 21) 清浄区域の室圧は,扉などで隣接する清浄度レベルの低い区域の室圧よりも高く設定すること.ただし,封じ込め施設の場合には,対象物質の拡散を防止し,かつ清浄度も維持できるよう室圧設定を工夫すること. |
6..最終滅菌法による医薬品の製造区域 6.2.2 空気 1)空気の清浄度レベルの異なる区域の間には、室間差圧を設定し、管理及び監視を行うこと。 2)滅菌前製品のバイオバーデンを管理する上で特に重要と考えられる差圧については、監視測定を常時行うこと。さらに、異常時に備えて警報システムを備えることが望ましい。 |
第5 章 製造 製造における交叉汚染の防止 5.21 品質リスクマネジメントのプロセスの結果に基づいて、交叉汚染に係るリスクを管理するため求められる技術的措置及び組織的措置の範囲を確定させること。 技術的措置及び組織的措置には以下が含まれ得るが、これらに限らない。 技術的措置 x . 潜在的な浮遊性汚染物質を特定区域内に封じ込めるよう、エアロック及び気圧カスケードを適切に用いる。 |
4 建物 4.4 無菌製品の製造には、クリーンルーム/ 区画について4つの清浄度等級がある。 グレードB: 無菌操作法による調製・容器充填作業のための、(アイソレータでない場合に)グレードAのバックグラウンドのクリーンルームである。差圧が連続してモニターされていること。 |
4.14 クリーンルームは、全ての作業条件下で、より低い等級のバックグラウンド環境に比して陽圧及び/又は気流を保持するフィルタ処理された給気が供給されていること、また、その給気は当該区域を効果的に換気するものであること。異なる清浄度等級の隣接する部屋には、少なくとも10パスカル(ガイダンス値) の気圧差があること。重要区域の保護には、特別な注意を払うこと。ある種の原材料(例: 病原性、高毒性若しくは放射性の生成物、又は生きたウイルス若しくは細菌性の原材料) を封じ込めることが必要な場合において、給気及び気圧に関する推奨事項を修正しなければならないことがあり得る。当該修正には、危険物質が周囲の区域を汚染するのを防止する陽圧又は陰圧がかかったエアロックが含まれうる。 | 4.16 気圧差の表示器が、クリーンルーム及び/又はアイソレータとそのバックグラウンドとの間に取り付けられていること。設置箇所及び気圧差の重要度が、CCSの中で検討されていること。重要と特定された気圧差は、連続してモニターされ且つ記録作成されていること。警報システムが整っていて、空気供給に不具合及び気圧差の低下(重要と特定された箇所について設定された限度値を下回る) があれば作業者に即時に表示され警報を発するようになっていること。 | (続き) 警報シグナルを評価なしに無効としてはならず、警報シグナルが発せられたときにとるべき手立てを概説する手順が用意されていること。警報の遅延を設定する場合には、CCSにおいて評価し、妥当性を示すこと。その他の気圧差は、規則的な間隔でモニターされ且つ記録されていること。 |
用語解説 汚染制御ストラテジー(CCS) - 微生物、エンドトキシン/発熱性物質及び微粒子を制御する計画一式であり、現行の製品及び工程の理解から導き出され、工程性能及び製品品質を保証するもの。当該制御には、原薬、添加剤並びに薬剤の原材料及び構成物、施設及び設備の操作条件、工程内管理、最終製品の規格、並びに付随するモニタリング及び管理の頻度及び方法に関連するパラメータ及び特性が含まれ得る。 |
パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.7 無菌製品を加工するには、陽圧管理区域を使用すること。ただし、封じ込めの理由から、病原体が露出する特定区域内の陰圧管理は許容される。特定のリスクのある原材料( 例: 病原体) の無菌処理に陰圧管理区域又はB S C を使用する場合には、適切な清浄グレードの陽圧管理ゾーンをその周囲に設けること。それらの気圧カスケードを明確に規定するとともに、アネックス1 に規定されているような適切なアラーム設定で継続的にモニターすること |
3.11 閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセス( 例: 添加剤、培地、緩衝液、ガスを添加する間のプロセス) では、適切な環境条件を適用すること。無菌操作については、アネックス1 に準拠したパラメータ( すなわち、グレードB を背景環境とするグレードA ) を適用すること。環境モニタリングのプログラムには、非微生物汚染、微生物汚染及び空気差圧の試験及びモニタリングを含めること。モニターする場所は、Q R M の原則を考慮して決定すること。モニタリングの検体数、量及び頻度並びにアラート及び対処の限度値は、Q R M の原則を考慮して適切であること。 | パートA . 一般的ガイダンス 建物及び設備 12. 無菌製品を加工するには、陽圧管理区域を使用すること。ただし、封じ込めの理由から、病原体が露出する特定区域内の陰圧管理は許容される。特にリスクのある原材料( 例: 病原体) の無菌処理に陰圧管理区域又は安全キャビネットを使用する場合には、適切な清浄グレードの陽圧管理ゾーンを周囲に設けること。それらの気圧カスケードを明確に規定するとともに、適切なアラーム設定を行い継続的にモニターすること。 |
無菌製造 24. 放射性粒子を封じ込めるため、製品が曝露されている区域の空気圧を、周辺区域よりも低くする必要がある場合がある。しかし、製品を環境汚染から保護することも必要である。これは例えば、気圧の壁として機能するバリア技術やエアロックを使用すれば可能であろう。 26. 放射性医薬品の生産に関しては、適切な差圧、気流の方向、空気の質を決定するために、リスク評価を適用できる。 |
Subpart C--Buildings and Facilities Sec. 211.42(c)Operations shall be performed within specifically defined areas of adequate size. There shall be separate or defined areas or such other control systems for the firm’s operations as are necessary to prevent contamination or mixups during the course of the following procedures: (10) Aseptic processing, which includes as appropriate: (iii) An air supply filtered through high-efficiency particulate air filters under positive pressure, regardless of whether flow is laminar or nonlaminar; |
IV. Buildings and Facilities C. Clean Area Separation (略) It is vital for rooms of higher air cleanliness to have a substantial positive pressure differential relative to adjacent rooms of lower air cleanliness. For example, a positive pressure differential of at least 10-15 Pascals (Pa) should be maintained between adjacent rooms of differing classification (with doors closed). |
(略) Maintaining a pressure differential (with doors closed) between the aseptic processing room and these adjacent rooms can provide beneficial separation. In any facility designed with an unclassified room adjacent to the aseptic processing room, a substantial overpressure (e.g., at least 12.5 Pa) from the aseptic processing room should be maintained at all times to prevent contamination. If this pressure differential drops below the minimum limit, it is important that the environmental quality of the aseptic processing room be restored and confirmed. |
(続き) The Agency recommends that pressure differentials between cleanrooms be monitored continuously throughout each shift and frequently recorded. All alarms should be documented and deviations from established limits should be investigated. |
Chapter 5: Production Prevention of cross-contamination in production 5.21 The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organisational measures required to control risks for cross-contamination. These could include, but are not limited to, the following: Technical Measures x. Appropriate use of air-locks and pressure cascade to confine potential airborne contaminant within a specified area; |
4 Premises 4.4 For the manufacture of sterile products there are four grades of cleanroom/zone. Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. |
4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of different grades should have an air pressure difference of a minimum of 10 Pascals (guidance value). Particular attention should be paid to the protection of the critical zone. The recommendations regarding air supplies and pressures may need to be modified where it is necessary to contain certain materials (e.g. pathogenic, highly toxic or radioactive products or live viral or bacterial materials). The modification may include positively or negatively pressurized airlocks that prevent the hazardous material from contaminating surrounding areas. | 4.16 Indicators of air pressure differences should be fitted between cleanrooms and/or between isolators and their background. Set-points and the criticality of air pressure differences should be considered within the CCS. Air pressure differences identified as critical should be continuously monitored and recorded. A warning system should be in place to instantly indicate and warn operators of any failure in the air supply or reduction of air pressure differences (below set limits for those identified as critical). | (続き) The warning signal should not be overridden without assessment and a procedure should be available to outline the steps to be taken when a warning signal is given. Where alarm delays are set, these should be assessed and justified within the CCS. Other air pressure differences should be monitored and recorded at regular intervals. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
PART A: GENERAL GUIDANCE SUPPLIMENTARY PROVISIONS TO PIC/S GMP GUIDE PART I CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Production Areas 3.7 Positive pressure areas should be used to process sterile products, but negative pressure in specific areas at the point of exposure of pathogens is acceptable for containment reasons. Where negative pressure areas or BSCs are used for aseptic processing of materials with particular risks (e.g. pathogens), they should be surrounded by a positive pressure clean zone of appropriate Grade. These pressure cascades should be clearly defined and continuously monitored with appropriate alarm settings as defined by Annex 1. |
3.11 Where processes are not closed and there is exposure of the product to the immediate room environment without a subsequent microbial inactivation process, (e.g. during additions of supplements, media, buffers, gasses, manipulations) appropriate environmental conditions should be applied. For aseptic manipulations parameters in line with Annex 1 (i.e. Grade A with Grade B background) should be applied. The environmental monitoring program should include testing and monitoring of non-viable contamination, viable contamination and air pressure differentials. The monitoring locations should be determined having regards to the QRM principles. The number of samples, volume, and frequency of monitoring, alert and action limits should be appropriate taking into account the QRM principles. | PART A: GENERAL GUIDANCE PREMISES AND EQUIPMENT 5. As part of the control strategy, the degree of environmental control of particulate and microbial contamination of the production premises should be adapted to the active substance, intermediate or finished product and the production step, bearing in mind the potential level of contamination of the starting materials and the risks to the product. The environmental monitoring programme should be supplemented by the inclusion of methods to detect the presence of specific microorganisms (i.e. host organism, yeasts, moulds, anaerobes, etc) where indicated by the QRM process. |
12. Positive pressure areas should be used to process sterile products but negative pressure in specific areas at the point of exposure of pathogens is acceptable for containment reasons. Where negative pressure areas or safety cabinets are used for aseptic processing of materials with particular risks (e.g. pathogens), they should be surrounded by a positive pressure clean zone of appropriate grade. These pressure cascades should be clearly defined and continuously monitored with appropriate alarm settings. | Sterile production Premises and equipment General 24. In order to contain radioactive particles, it may be necessary for the air pressure to be lower where products are exposed, compared with the surrounding areas. However, it is still necessary to protect the product from environmental contamination. This may be achieved by, for example, using barrier technology or airlocks, acting as pressure sinks. 26. For manufacture of radiopharmaceuticals a risk assessment may be applied to determine the appropriate pressure differences, air flow direction and air quality. |
Chapter 5: Production Prevention of cross-contamination in production 5.21 The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organisational measures required to control risks for cross-contamination. These could include, but are not limited to, the following: Technical Measures x. Appropriate use of air-locks and pressure cascade to confine potential airborne contaminant within a specified area; |
4 Premises 4.4 For the manufacture of sterile products there are four grades of cleanroom/zone. Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. |
4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of different grades should have an air pressure difference of a minimum of 10 Pascals (guidance value). Particular attention should be paid to the protection of the critical zone. The recommendations regarding air supplies and pressures may need to be modified where it is necessary to contain certain materials (e.g. pathogenic, highly toxic or radioactive products or live viral or bacterial materials). The modification may include positively or negatively pressurized airlocks that prevent the hazardous material from contaminating surrounding areas. | 4.16 Indicators of air pressure differences should be fitted between cleanrooms and/or between isolators and their background. Set-points and the criticality of air pressure differences should be considered within the CCS. Air pressure differences identified as critical should be continuously monitored and recorded. A warning system should be in place to instantly indicate and warn operators of any failure in the air supply or reduction of air pressure differences (below set limits for those identified as critical). | (続き) The warning signal should not be overridden without assessment and a procedure should be available to outline the steps to be taken when a warning signal is given. Where alarm delays are set, these should be assessed and justified within the CCS. Other air pressure differences should be monitored and recorded at regular intervals. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Sterile production Premises and equipment General 24. In order to contain radioactive particles, it may be necessary for the air pressure to be lower where products are exposed, compared with the surrounding areas. However, it is still necessary to protect the product from environmental contamination. This may be achieved by, for example, using barrier technology or airlocks, acting as pressure sinks. 26. For manufacture of radiopharmaceuticals a risk assessment may be applied to determine the appropriate pressure differences, air flow direction and air quality. |
4 Premises 4.4 For the manufacture of sterile products there are four grades of cleanroom/zone. Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. |
4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of different grades should have an air pressure difference of a minimum of 10 Pascals (guidance value). Particular attention should be paid to the protection of the critical zone. The recommendations regarding air supplies and pressures may need to be modified where it is necessary to contain certain materials (e.g. pathogenic, highly toxic or radioactive products or live viral or bacterial materials). The modification may include positively or negatively pressurized airlocks that prevent the hazardous material from contaminating surrounding areas. | 4.16 Indicators of air pressure differences should be fitted between cleanrooms and/or between isolators and their background. Set-points and the criticality of air pressure differences should be considered within the CCS. Air pressure differences identified as critical should be continuously monitored and recorded. A warning system should be in place to instantly indicate and warn operators of any failure in the air supply or reduction of air pressure differences (below set limits for those identified as critical). | (続き) The warning signal should not be overridden without assessment and a procedure should be available to outline the steps to be taken when a warning signal is given. Where alarm delays are set, these should be assessed and justified within the CCS. Other air pressure differences should be monitored and recorded at regular intervals. |
Glossary contamination control strategy (CCS). A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
4. Premises 4.4 Manufacturing facilities should normally be maintained at a positive pressure relative to the outside, to prevent the ingress of contaminants. Where facilities are to be maintained at negative pressures relative to the outside, special precautions should be taken to mitigate any risks (see (3)). 4.9 Containment, cleanliness and protection may be facilitated through, for example: ・ sufficient pressure differentials. 4.10 Detailed schematic diagrams should be maintained, indicating, for example, air supply and air return, room pressure differentials and airflow directions. |
7. Air filtration, airflow direction and pressure differentials 7.1 Where different products are manufactured at the same time, i.e. in different areas or rooms in a multiproduct manufacturing site, measures should be taken to ensure that dust cannot move from one room to another. Facility design and layout, appropriate levels of filtration, airflow direction and pressure differentials can assist in preventing cross-contamination. | 7.4 The pressure differential between areas in a facility should be individually assessed according to the products handled and level of protection required. The pressure differential and the direction of airflow should be appropriate to the product and processing method used, and should also provide protection for the operator and the environment. 7.5 The pressure differential should be designed so that the direction of airflow is from the clean area, resulting in dust containment, for example, from the corridor to the cubicle. 7.6 The limits for the pressure differential between adjacent areas should be such that there is no risk of overlap in the defined dynamic operating ranges. |
7.7 Normally, for rooms where dust is liberated, the corridor should be maintained at a higher pressure than the rooms and the rooms at a higher pressure than atmospheric pressure. (For negative pressure facilities refer to WHO good practices for pharmaceutical products containing hazardous substances (3), for guidelines and design conditions.) Room pressure differential indication should be provided. This may be by pressure gauges or suitable electronic systems such as EMS or BMS. Where pressure indication gauges are provided, these should have a range and graduation scale that enables them to be read to an appropriate accuracy. The normal operating range, alert and action limits should be defined and displayed at the point of indication or EMS/BMS. | (続き) Room pressure should be traced back to representative ambient pressure (by summation of the room pressure differentials), in order to determine the actual absolute pressure in the room. 7.11 Where airlocks are used, the pressure differentials selected should be appropriate. When selecting room pressure differentials, transient variations, such as machine extract systems and their impact, should be taken into consideration. |
4. Premises 4.1 Premises design The infiltration of contamination from outside air should be minimized by the use of appropriate filtration, room pressure differentials and airlocks. Manufacturing facilities should normally be maintained at a positive pressure relative to the outside, to limit the ingress of contaminants. Where facilities are to be maintained at negative pressures relative to the ambient pressure, special precautions should be taken to avoid ingress and egress of contaminant. |
(続き、略) Where necessary, air locks, change rooms and pass-through hatches may be considered and provided with effective ventilation and filtered air. Special attention should be given to door design, as gaps between doors and floors, doors opening into low-pressure areas, and sliding doors can result in changes in the pressure differential between areas. An interlocking system and a visual and/or audible warning system may be used, where required, to prevent opening of more than one door at a time where required. |
4.2 Weighing/dispensing and sampling areas
A room for weighing (e.g. dispensing of materials) should be of appropriate design (for examples, see Figs A2.1 and A2.2). (略) The HVAC system for such areas should ensure that the areas have at least the same area classification as other production areas where materials and products are exposed to the environment, logical flow of material and personnel, and an appropriate number of AHUs, as well as appropriate pressure differentials, containment, dust control, and rate of air exchange. |
(続き、略) To further support containment, consideration may also be given to having material airlocks (MALs) and personnel airlocks (PALs), where needed, for entry and exit of processing areas (for an example, see Fig. A2.6). Appropriately designed airlocks can assist in ensuring containment. Additional controls, such as pressure differentials between areas, an appropriate number of air changes in an area, and sufficient filtration of air, should be in place. The use of airlocks assists in ensuring containment; however, other means may be considered to achieve this objective, such as closed systems and pressure gradients between adjacent areas. |
(続き、略) Washing areas should be designed and used in such a manner that equipment and components will not be re-contaminated after cleaning. The system supplying and extracting air from the area(s) should be suitably designed to ensure that this objective is achieved. Principles that may be considered include (but are not limited to) filtration of air, pressure differentials between areas, air changes per hour and airflow directions (for an example, see Fig. A2.7). |
5. Design of HVAC systems and components 5.1 Containment Manufacturers should ensure that appropriate measures are taken to contain product dust in a manufacturing area, thus preventing or minimizing the risk of contamination of other areas and possible cross-contamination. In some cases, it may be advisable to have airlocks or pass-through hatches between rooms or areas. In addition, sufficient dilution, pressure differentials (recommended minimum values of 5 Pa) and airflow directions can further support containment in an area. |
5.3 Automated monitoring systems The performance of the HVAC system achieving and maintaining the desired results for parameters such as temperature, relative humidity, airflow and pressure differential should be carefully controlled and monitored. |
7. Air filtration, airflow direction and pressure differentials The pressure differential should be of sufficient magnitude to ensure containment and prevention of flow reversal, but should not be so high as to create turbulence problems. It is suggested that pressure differentials of between 5 Pa and 20 Pa be considered. Where the design pressure differential is too low and tolerances are at opposite extremities, a flow reversal can take place. There should be no risk of overlap in the acceptable operating range, for example, 5 Pa to 15 Pa in one room and 15 Pa to 30 Pa in an adjacent room, resulting in failure of the pressure cascade (for examples, see Fig. A2.10). | (続き) The upper and lower limits for pressure differentials between areas in a facility should be defined by the manufacturer. Where there are interleading rooms, the limits should be appropriate to ensure that there is no overlap in actual values, as this may result in loss in pressure differential between areas and even reversal of air flow. |
(続き) The upper and lower limits for pressure differentials between areas in a facility should be defined by the manufacturer. Where there are interleading rooms, the limits should be appropriate to ensure that there is no overlap in actual values, as this may result in loss in pressure differential between areas and even reversal of air flow. |
7.1 Airlocks Airlocks with different pressure cascade regimes include the cascade airlock, sink airlock and bubble airlock: ・cascade airlock: higher pressure on one side of the airlock and lower pressure on the other (for an example, see Fig. A2.11); ・ sink airlock: lower pressure inside the airlock and higher pressure on both outer sides (for an example, see Fig. A2.12); ・ bubble airlock: higher pressure inside the airlock and lower pressure on both outer sides (for an example, see Fig. A2.13). |
(続き) Fig. A2.11 Example of a cascade airlock: in most cases, the internal pressure of the airlock is not critical; the pressure differential between the two outer sides is the important criterion Fig. A2.12 Example of a sink airlock Fig. A2.13 Example of a bubble airlock Note: The diagrams above and the differential pressures shown here are for illustration purposes only. Pressures indicated in these examples are absolute pressures, whereas the local pressure indication would most likely be the pressure differential from room to room. |
9. Premises 9.9 The HVAC system and pressure cascade design for the different areas should be appropriately designed and maintained, in order to minimize the risk of product contamination and to protect personnel from the risks of radiation exposure. The pressure differentials should be controlled, monitored and recorded. Appropriate controls should be put in place to promote the containment of radioactivite gases and vapours. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬部外品 以下の要件があります。 ・ 無菌操作を行う区域の適切な差圧管理。 *「適切な差圧」の具体的数値表記はありません. |
対象医薬品:無菌操作法による特定生物由来医薬品等 以下の要件があります。 ・ 無菌操作を行う区域の適切な差圧管理。 ・病原性を持つ微生物等を取り扱う区域の陰圧管理。 *「適切な差圧/陰圧」の具体的数値表記はありません。 |
対象医薬品:無菌操作法による特定生物由来医薬品等 以下の要件があります。 ・ 無菌操作を行う区域の適切な差圧管理。 ・病原性を持つ微生物等を取り扱う区域の陰圧管理。 *「適切な差圧/陰圧」の具体的数値表記はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・清浄区域における適切な室間差圧の設定と監視。 ・隣接する清浄室の室圧設定。 *「適切な差圧」の具体的数値表記はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌操作区域とその他の支援区域間の差圧設定・監視・管理について推奨差圧10〜15Pa。 ・その他の支援区域内の清浄度の異なる区域間における適切な差圧設定。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌性確保の上で重要な差圧のモニタリング。 ・製造作業中の差圧変動について。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・清浄区域における適切な室間差圧の設定と監視。 ・隣接する清浄室の室圧設定。 *「適切な差圧」の具体的数値表記はありません。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・清浄度の異なる区域間における適切な差圧設定。 ・バイオバーデン管理に重要な差圧についての常時モニタリング。 *「適切な差圧」の具体的数値表記はありません。 |
対象医薬品: 全般 以下の要件があります。 ・製造における交叉汚染の防止のため、潜在的な浮遊性汚染物質を特定区域内に封じ込めるよう、圧力カスケードを用いる。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードBにおける差圧の連続モニタリング。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 用語ついての補足:「差圧」は「Air pressure differences」 を訳しており、全体を示す箇所に使用しており、「気圧差」は同じ「air pressure differences」の訳として数値を扱う箇所に使用しています。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの陽圧について。 ・異なる清浄度グレードで隣接する室との差圧の推奨値(少なくとも10Pa)。 ・封込めが必要な場合(例:病原性、高毒性若しくは放射性の生成物、又は生きたウイルス若しくは細菌性の原材料)は、陽圧および差圧に関する推奨事項を修正すること、およびその修正には陽圧および陰圧のエアロックが含まれること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルーム間、アイソレーターと周囲環境間の差圧計について。 ・差圧の設定値と重要性とCCSについて。 ・重要差圧の連続モニタリング、警報。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 *【誤記訂正】厚生労働省通知の和訳ではここを ”CSS” と表記されていますが、PIC/Sの原文は ”CCS”であるため、誤記です。表は正しい ”CCS”を記載。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・重要差圧の警報遅延の設定は、CCSによる評価と妥当性を示すこと。 ・警報の発報時の停止には、手順の準備が必要。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCSの定義。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・無菌製品の加工区域における陽圧・陰圧の設定および室圧カスケードの設定・管理。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセスのプロセスの差圧の試験と環境モニタリング。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 以下の要件があります。 ・無菌製品の加工区域における陽圧・陰圧の設定および室圧カスケードの設定・管理。 |
対象医薬品:放射性医薬品のうち無菌製品 以下の要件があります。 ・放射性粒子の封じ込めと製品を環境汚染から保護するためのバリア技術やエアロックを用いた差圧設定。 ・適切な差圧設定におけるリスク評価の適用。 *「適切な差圧」の具体的数値表記はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌製造における陽圧について。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・隣接した異なる清浄度設定間の差圧について例として、少なくとも10ー15Pa。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌操作区域と隣接した異なる清浄度設定間の差圧推奨値(少なくとも12.5Pa)。 ・清浄度規定のない隣接室との室間差圧についての差圧推奨値および下限値を下回った場合の対応について。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・差圧の連続モニタリング・記録・アラート・逸脱について。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第5 章 製造 製造における交叉汚染の防止 5.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.7. と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA : 一般的ガイダンス 第3章 建物及び設備 建物 製造区域3.11と同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤)の製造 パートA : 一般的ガイダンス 建物及び設備 5と同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤)の製造 パートA : 一般的ガイダンス 建物及び設備 12と同じ内容となります。 |
対象医薬品:放射線医薬品のうち無菌医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 24, 26と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第5 章 製造 製造における交叉汚染の防止 5.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:放射性医薬品のうち無菌製品 別紙(2) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 無菌製造 24. 、26. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・ 陽圧・陰圧の設定。 ・封じ込め、清浄度や保護の場合の十分な差圧。 ・気流方向などを精密に示した関連図について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・施設内で異なる製品を同時に製造する場合の差圧。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・製造する製品や製造方法、作業者や環境の保護における差圧の設定。 ・清浄度のレベルにおける差圧方向。 ・差圧の許容値・変動幅について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・部屋の圧力を大気圧よりも高く維持し、塵が発生する部屋では、廊下の圧力を部屋よりも高くすること。 ・差圧ついては差圧計もしくはEMS/BMSにおいて表示すること。 用語ついての補足: ・BMS: Building Management Systemの略で、ビル管理システムのこと。 ・EMS: Environmental Monitoring System の略で、環境モニタリングシステムのこと。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・基準圧との関係について。 ・エアロックにおける差圧の変動について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・室間差圧による汚染防止策について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・扉の仕様(床面とのすき間、開き方向、引き戸)と差圧への影響について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・秤量/小分けおよびサンプリング区域の適切な室間差圧。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・封じ込め目的でのエアロック設置を含む、隣接するエリアの差圧との圧力勾配について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・洗浄室エリアの空調について清浄化済みの装置や部品が再汚染しないよ差圧を設定すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・封じ込めにおいて室間差圧(推奨値最小5 Pa)及び気流方向の適切な対応をとること |
対象医薬品:非無菌医薬品 以下の要件があります。 差圧を含む空調システムの性能を維持するために、モニタリングステムにより、注意深く、管理・監視すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・室間差圧推奨値( 5 〜 20 Pa)。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・差圧が小さくかつそれぞれの室の室圧許容幅が大きい場合、室圧逆転が発生するため、許容幅がオーバーラップしないよう設定すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・差圧計測においては、機器許容誤差により検知できないまま気流の逆流が発生しないようにすること。 ・隣接する室間で(室圧の)許容幅が重ならいように設定すること。 ・それぞれの室圧は共通の基準圧点から取ること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・カスケード型エアロック、シンク型エアロック、およびバブル型エアロックそれぞれの差圧の設定。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・カスケード型エアロック、シンク型エアロック、およびバブル型エアロックそれぞれの図示。 ・各所において設置する室圧計と室間の差圧計について。 |
対象医薬品:放射線医薬品 以下の要件があります。 ・製品汚染を最小にし、作業者が放射性物質に曝露しないような室圧カスケード。 ・放射性ガスや蒸気の封じ込めにおける室間差圧。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | ― | ― | ― | 欧州 | 欧州 | ― | ― | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 分 類 | 医薬・生活衛生局監視指導・麻薬対策課長 事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令の一部改正に ついて | 医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令の一部改正に ついて | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2A ヒト用先端医療医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2B ヒト用生物学的医薬品( 原薬及び製剤) の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 2A MANUFACTURE OF ADVANCED THERAPY MEDICINAL PRODUCTS FOR HUMAN USE |
EU Guidelines for Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part 1 |
EU Guidelines for Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2021年 | 2011年 | 2012年 | 2020年 | 2025年 | 2022年 | 2022年 | 2004年 | 2021年 | 2023年 | 2023年 | 2020年 | 2022年 | 2022年 | 2019年 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 記載内容 |
第3 逐条解説 ≪第1節通則 (第3条の3ー第 20 条)≫ 11.第8条(手順書等)関係 (1)製造・品質関連業務を適正かつ円滑に行う ため、製造所ごとに 所定の手順について記載した文書の作成等について規定するものであること。 (略) ①第8条 第1項第1号関係 ウ構造設備及び職員の衛生管理に関する手順 として、その製造所における製造工程等に応じて、次に掲げる手順のうち該当するもの について記載するものであること 。 <無菌医薬品 区分製造所の場合> ㋒作業室又は作業管理区域の清浄度の維持管理に関する手順 |
≪第1節通則 (第 32 条ー第 48 条)≫ 46.第 36 条(手順書)関係 (1)第36 条第1号関係 ③構造設備及び職員の衛生管理に関する手順 として 、その製造所における製造工程等に応じて、次に掲げる手順のうち該当するものについて記載するものであること 。 <施行規則第 25 条第 2 項第 1 号又は第 35 条第 2 項第 1号の区分の製造所(以下「無菌医薬部外品 区分製造所」という。) の場合> (イ) 作業室又は作業管理区域の清浄度の維持管理に関する手順 |
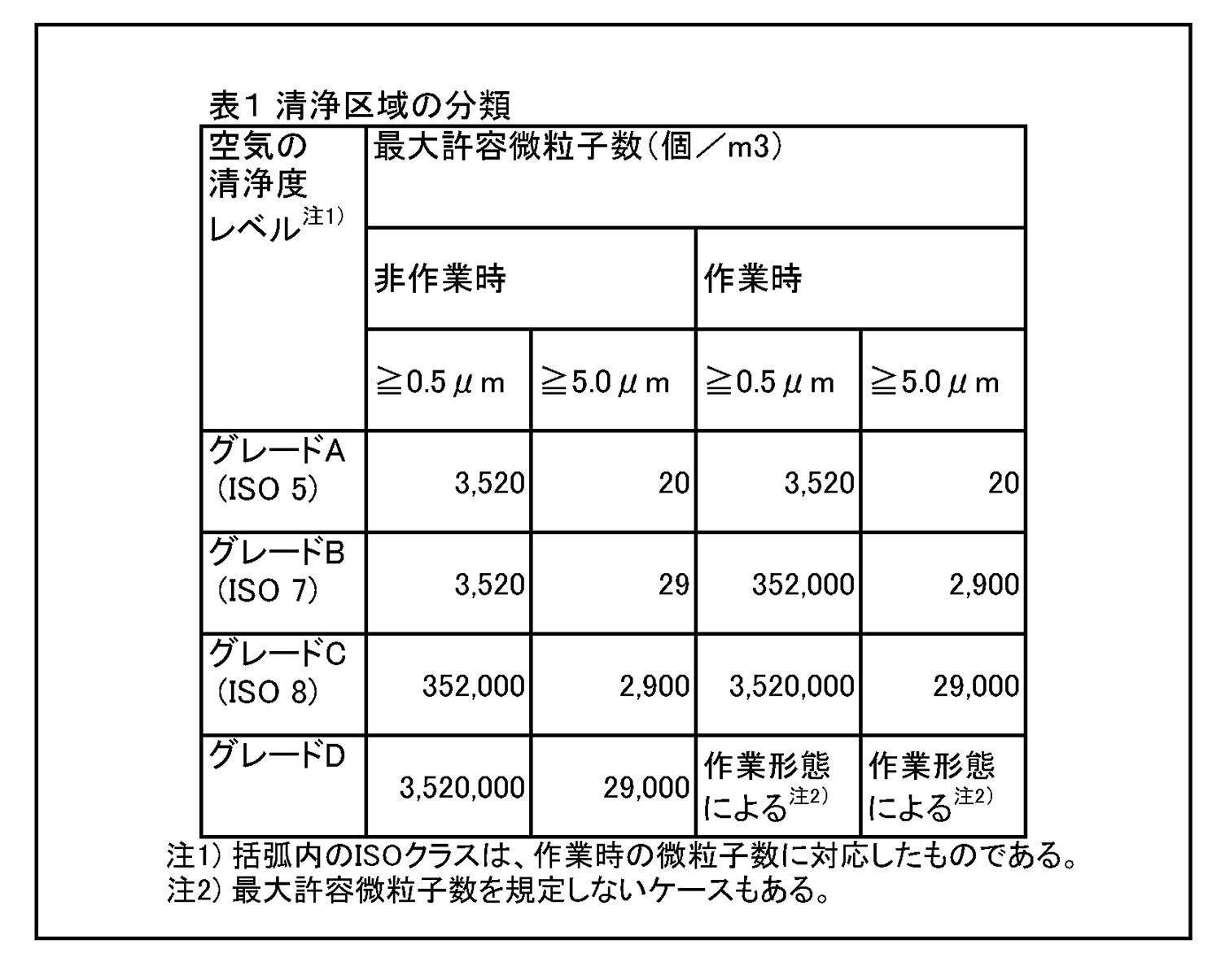
7.無菌医薬品に係る製品の作業所 7.1 清浄度レベルによる作業所の分類 一般的に各区域の清浄度レベルは、環境空気の単位体積当たりに含まれる粒径0.5μm以上の浮遊微粒子数によって表される。また、粒径5.0μm以上の浮遊微粒子数は定期的に測定し、傾向分析を行うことにより、環境条件の劣化を早期に発見するための有効な管理項目となる。各区域に要求される空気の清浄度レベルを表1に示す。 |
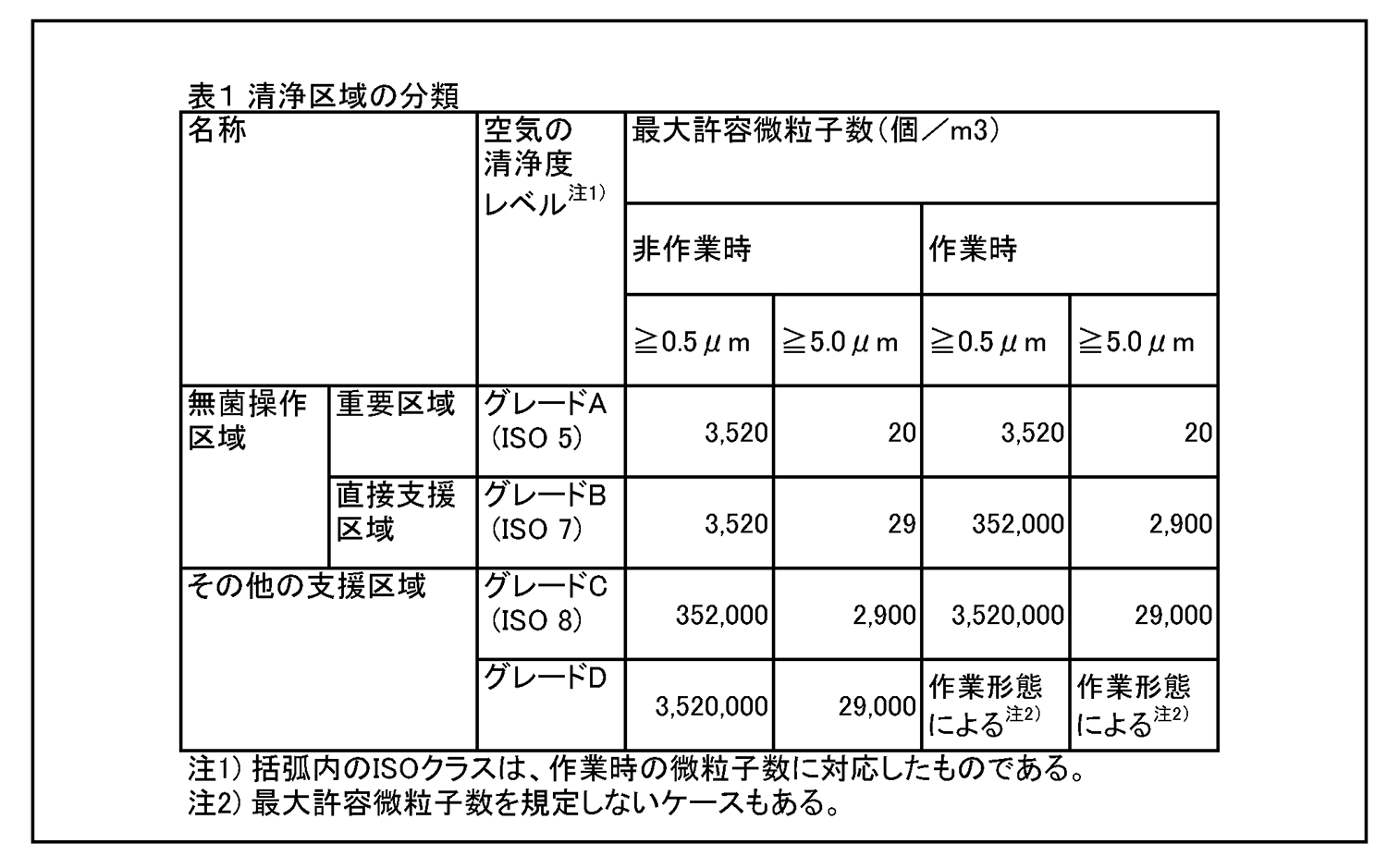 |
6.最終滅菌法による医薬品の製造区域 6.1 清浄度レベルによる作業所の分類 一般的に各区域の清浄度レベルは、環境空気の単位体積当たりに含まれる粒径0.5μm以上の浮遊微粒子数によって表される。また、粒径5.0μm以上の浮遊微粒子数を定期的に測定し、傾向分析を行うことにより、環境条件の劣化を早期に発見するための有効な管理項目となる。各区域に要求される空気の清浄度レベルを表1に示す。 最終滅菌法による無菌医薬品に係る製品の作業所は、浮遊微粒子及び微生物による汚染程度が所定限度内に維持されるよう管理された区域であり、その作業内容により、充てん・閉そく区域、直接支援区域、その他の支援区域に分類される。 |
 |
第3 章 建物及び設備 製造区域 3.7. 作業の流れ及び必要な清浄度レベルに応じた論理的な順序で連結した区域において製造が行われるよう、建物を配置することが望ましい。 |
4 建物 4.4 無菌製品の製造には、クリーンルーム/ 区画について4つの清浄度等級がある。 グレードA:高リスク作業のための重要区画( 例: 無菌操作ライン、容器充填区画、止栓ボウル、開口状態の1次包装又はファーストエアの保護下で無菌接続を行う区画) 。通常、当該条件は、局所的な気流保護(RABS又はアイソレータ内での一方向気流作業台等)によって得られる。一方向気流が保たれていることが、グレードA区域全体にわたって実証され且つ適格性評価されていること。 |
(続き) 作業者によるグレードA 区域内への直接の介入操作( 例: バリア技術又は手袋ポート技術での保護されていない介入操作)を最小化するように、建物、設備、工程及び手順が設計されていること。 |
(続き) グレードB: 無菌操作法による調製・容器充填作業のための、(アイソレータでない場合に)グレードAのバックグラウンドのクリーンルームである。差圧が連続してモニターされていること。アイソレータ技術を用いている場合には、グレードBより低い等級のクリーンルームを検討し得る(4.20節を参照) 。 グレードC及びD:無菌操作法により容器充填済みの無菌製品の製造における比較的重要でない段階に用いられ、又はアイソレータのバックグラウンドとして用いられるクリーンルームである。また、最終滅菌法による製品の調製/容器充填作業に用いられることもある。(最終滅菌作業に関する特別な詳細については8項を参照) 。 |
4.12 (略) エアロックをフィルタ処理された空気で効果的に換気して、クリーンルームの等級が維持管理されていることを確保にすること。エアロックの最終段階は、「非作業時」の状態において、そこからつながるクリーンルームと同じ清浄度等級( 生菌数及び総微粒子量) になっていること。グレードB区域を入退室する際には、別々の更衣室を使うことが望ましい。それが実践的でない場合には、(入室/退室) 作業の時間を別にする手順を検討すること。CCSで汚染のリスクが高いことが示されている場合には、製造区域の入退室で別々の更衣室が使われること。エアロックは、以下のように設計されていること: |
(続き) i.人員用のエアロック: 人員の立入りに使われる、清浄度が次第に高くなる区域( 例:グレードD区域からグレードC区域へ、続いてグレードB区域へ)。一般に手洗い施設は、更衣室の第一段階にのみ設けることとし、グレードB区域に直接つながっている更衣室内にあってはならない。 ii.原材料用のエアロック: 原材料及び設 備の搬送に使われる。 ・承認を受けたリストに収載されている原材料及び設備であって搬送工程のバリデーションの際に評価済みであるもののみが、グレードA又はグレードBの区域内へエアロック又はパススルーハッチを通して搬入されていること。 |
(続き) 設備及び原材料(グレードA区域内で用いようとするもの) がグレードB区域を通過する際に保護されていること。承認を受けていない物品で搬送を要するものがあれば、例外として事前承認を受けること。 リスクの適切な評価及び軽減措置は、その製造業者のCCSに則って適用され且つ記録作成されていること、また、品質保証担当によって承認を受けた特別な消毒及びモニタリングのプログラムを含めること。 |
(続き) ・パススルーハッチは、 例えば、有効なフィルタ処理された給気で効果的に換気することで) より高い等級の環境を保護するように設計されていること。 ・より低い等級の区域又は等級分けされていない区域からより高い等級の区域への原材料又は設備の搬送は、CCSに沿ってリスクに相応した清浄化及び消毒の対象となっていること。 |
バリア技術 4.20 アイソレータ又はRABSのバックグラウンド環境は、汚染の伝播するリスクが最小化されていることを確保するものであること。 i.アイソレータ a. 開口式アイソレータのバックグラウンド環境は一般に、最低限グレードC相当であること。閉鎖式アイソレータのバックグラウンド環境は、最低限グレードD相当であること。バックグラウンドの等級分けの決定は、リスク評価に基づくものであり、且つCCSにおいて妥当性が示されているものであること。 ii.RABS 無菌操作用のRABS のバックグラウンド環境は最低限グレードB相当であること、また、気流パターンの検討試験を行って、介入操作(該当する場合にはドアを開くことを含む)の際に空気が入ってこないことを実証すること。 |
クリーンルーム及び清浄空気設備の適格性評価 4.24クリーンルーム及び清浄空気設備は、アネックス15の要求事項に準拠する方法を用いて適格性評価されていること。クリーンルームの適格性評価(等級分けを含む) は、運用時の環境モニタリングと明確に区別されていること。 |
4.26 クリーンルームの等級分けは、クリーンルームの適格性評価の一部であり、また、微粒子の総濃度を測定することでクリーンルーム又は清浄空気設備の仕様に対する空気清浄度のレベルを評価する方法である。工程又は製品品質へのインパクトを回避するためには、等級分け作業が予定を組んで行われていること。例えば、導入時の等級分けがシミュレートされた作業の際に行われ、再等級分けがシミュレートされた作業又は無菌操作プロセスシミュレーション(APS) の際に行われていること。 | 4.27 クリーンルームの等級分けには、0.5μm及び5μm以上の微粒子の総量を測定すること。この測定は、表1に規定された限度値に準拠して、非作業時及びシミュレートされた作業時の両方で行われるものであること。 |
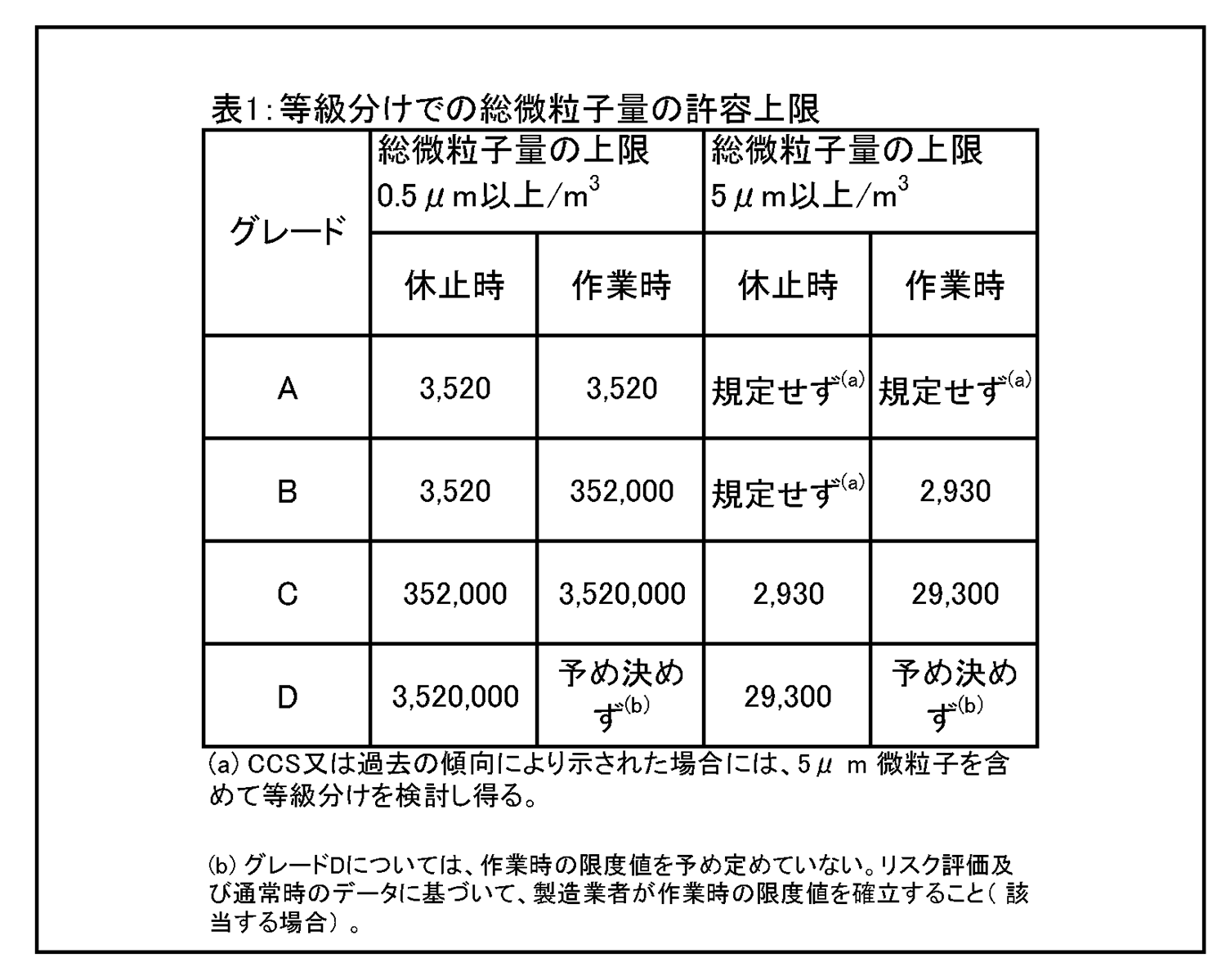 |
4.28 クリーンルームの等級分けについて、検体採取箇所の最低限の数及び位置決めがISO14644パート1に示されている。無菌操作区域及びそのバックグラウンド環境(それぞれグレードA及びグレードBの区域) については、追加の検体採取箇所を検討すること、また、容器充填部位及び容器密栓補充ボウル部等の重要工程区域は、全て評価すること。重要操作箇所は、文書化されたリスク評価及び当該区域内で行われる工程・作業についての知識によって決定すること。 | 4.29 クリーンルームの等級分けは、「非作業時」及び「作業時」の状態において行うこと。 i.「非作業時」状態の定義は、HVACを機能させることを含め全てのユーティリティの据付けが完了しており、主要な製造設備が規定どおりに据え付けられているが稼働しておらず、且つ室内に人員がいない状態である。 ii.「作業時」状態の定義は、クリーンルームの据付けが完了して、HVACシステムが完全に稼働中で、設備が据え付けられて製造業者の定めた稼働モードで機能しており、最大限の数の人員が居合わせて通常時の運用作業を実行している又はシミュレートしている状態である。 |
(続き) iii.「非作業時」状態について上記表1 中に示した総微粒子量の限度値は、作業及びラインクリアランス/ 清浄化が完了した「クリーンアップ」期を経た後に達成されるものであること。「クリーンアップ」期( ガイダンス値として20分未満)は、当該クリーンルームの適格性評価の際に決定され、文書化されていること、また、適格性評価された清浄度の状態が作業の際に途絶えたときには、復旧手順を厳守すること。 |
8 製造及び特有の技術 最終滅菌法による製品 8.1 微生物、エンドトキシン/発熱性物質及び微粒子のリスクを限定するため、構成物及び原材料の準備作業は最低でもグレードDクリーンルーム内で行い、製品が滅菌に適した状態となるようにすること。製品に微生物汚染のリスクが高い又は通常でない場合( 例:製品が微生物生育を活発にする場合、製品を容器充填前に長期間保持しておく必要がある場合、又は製品が殆ど密閉槽内で処理されない場合) には、準備作業は最低でもグレードC環境中で行うこと。 |
(続き) 軟膏剤、クリーム剤、懸濁化剤及び乳化剤の調製作業は、最低でもグレードC環境中で最終滅菌前に行うこと。最終滅菌法による動物用医薬品に関する特定のガイダンスは、GMPガイドラインのアネックス4 に示されている。 8.3 最終滅菌のための製品の容器充填は、少なくともグレードC環境中で行うこと。 8.4 例えば、容器充填作業がゆっくりである、容器の口が広い又は容器を閉じる前に数秒間以上おく必要がある等、製品が環境から通常でない汚染のリスクにおかれることがCCSで同定される場合には、最低でもグレードCバックグラウンドのグレードAにおいて当該製品を容器充填すること。 |
8.6 種々の清浄度等級において行われる作業の具体例を、表3 に示す。 |
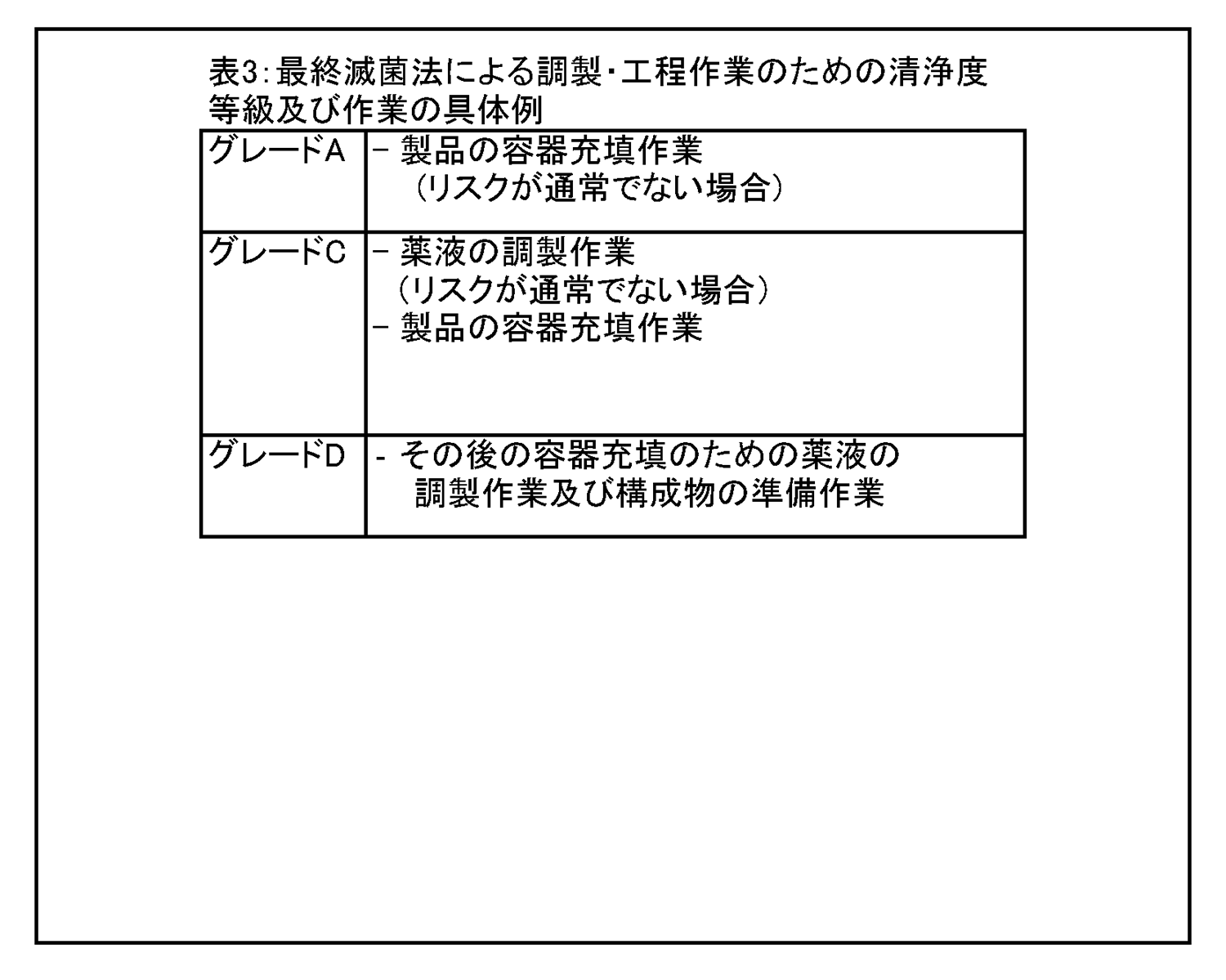 |
無菌操作法による調製・工程作業 8.9 グレードAへの重大な介入操作の必要性を減らして汚染のリスクを最小化するためには、なるべくRABS、アイソレータその他システムの設備を用いることを検討すること。工程をロボット化及び自動化して、直接人が重大な介入操作を行わないようにすることも検討し得る( 例: 乾熱処理トンネル、自動化された凍結乾燥機搬入、定置滅菌) 。 8. 10種々の環境清浄度等級において行われる作業の具体例を、表4に示す。 |
 |
8.12 製品に直接的又は間接的に接触する滅菌済みの設備、構成物及び付属品の開封作業、組立作業及び準備作業は、無菌操作工程として扱い、グレードBバックグラウンドを有するグレードA内で行うこと。容器充填ラインの始動準備及び無菌製品の容器充填作業は、無菌操作工程として扱い、グレードBバックグラウンドを有するグレードA内で行うこと。アイソレータを用いる場合には、そのバックグラウンドが4.20節に準拠すること。 | 8.13 軟膏剤、クリーム剤、懸濁剤及び乳剤等の無菌製品の調製作業及び容器充填作業は、その製品及び構成物が環境中に露出し、その製品が(滅菌グレードフィルタで) 濾過されず又は最終滅菌されないときには、グレードBバックグラウンドを有するグレードA 内で行うこと。アイソレータ又はRABSを用いる場合には、そのバックグラウンドが4 . 2 0 節に準拠すること。 | 8.14 無菌接続は、グレードBバックグラウンドを有するグレードA内で行うこと(なお、続いてその場で滅菌する、又は隣接環境からの潜在的汚染を最小化する組込み式無菌接続器具で行うときには、この限りでない) 。組込み式無菌接続器具は、汚染のリスクを軽減するように設計されていること。 アイソレータを用いる場合には、そのバックグラウンドが4.20節に準拠すること。無菌接続が適切に評価され、その有効性が検証されていること。組込み式無菌接続器具に関する要求事項については、8.129節及び8.130節を参照。 |
9 環境及び工程のモニタリング 9.9 生菌粒子及び総微粒子量のモニタリングの結果に対して、適切な警報基準値及び処置限度値が設定されていること。総微粒子量の処置限度値の上限は表5に記載されている、また、生菌粒子の処置限度値の上限は表6に記載されている。 |
9.12 グレードC及びDのクリーンルームについての作業中のモニタリングは、適格性評価の際に収集されたデータ及び通常時のデータに基づいて行って、効果的な傾向分析ができるようにすること。警報基準値及び処置限度値についての要求事項は、行われる作業の性質により異なってくる。処置限度値は、表5及び表6に掲げられた値よりも厳格になり得る。 環境モニタリング ー 総微粒子量 9.15 グレード毎の浮遊微粒子汚染の環境モニタリングの限度値を、表5に示す。 |
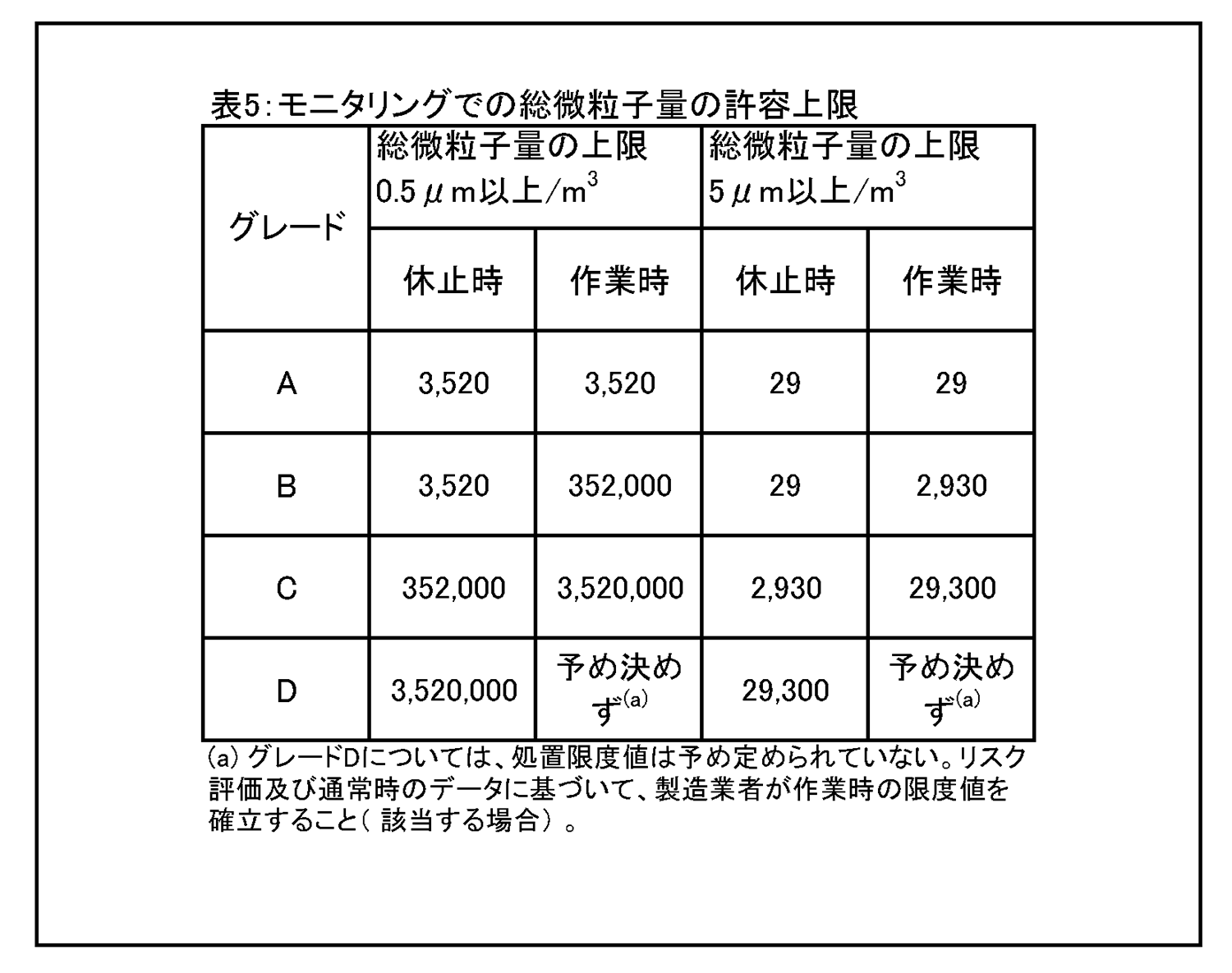 |
注1: 「非作業時」状態について表中に示されている微粒子限度値は、作業の完了後に無人状態で、適格性評価の際に定められた短時間の「クリーンアップ」期( ガイダンス値として20分未満) を経た後に達成されるものであること(4.29節を参照) 。 注2:グレードA 中においてマクロ粒子( 特に5μm以上のもの) 計数が時折表示される場合は、電気的なノイズ、迷光、偶然一致した減損等による誤計数であると考えてよい。ただし、低レベルで連続した又は規則的な計数があるときには、汚染事象の可能性を示唆していることがあり得るので調査すること。そうした事象は、室内空気供給のフィルタ処理システムの初期不具合、設備不具合を示しているおそれがあり、又は機械の始動準備及び通常時の作業の際の杜撰な慣行を診断することにもなり得る。 |
用語解説 アイソレータ ー内部に再現性のあるバイオ除染を行うことが可能な筐体で、グレードA条件に合致する内部作業区画を有し、その内部を毀損することなく外部環境(例:クリーンルームの周囲の空気及び人員)から持続的に隔離状態にするもの。アイソレータには主要な2つの種類がある。 i. 閉鎖式アイソレータシステムは、周囲環境への開放部を用いずに、補助的設備への無菌接続部を介して原材料を搬送することにより、アイソレータの内部が外から汚染されないようにする。閉鎖システムは作業の間、密封状態に保たれる。 |
(続き) ii. 開口式アイソレータシステムは、1以上の開口部があり、作業の際に連続的又は半連続的に原材料を出し入れできるように設計されている。開口部は、外からの汚染物質がアイソレータに入り込まないようにする造り(例:持続的な過圧を用いる)になっている。 |
用語解説 アクセス制限バリアシステム(RABS) ー所定の空気品質条件(無菌操作にはグレードA)に合致する閉鎖された(だが完全密封ではない)環境を提供するシステムで、堅牢な壁で囲われた筐体と一体化した手袋を用いて、周囲のクリーンルーム環境からその内部を分離するシステム。RABSの内表面は殺芽胞剤で消毒及び除染される。作業者は、手袋、半身スーツ、RTPその他の一体化された搬送ポートを使用して、RABS内部に操作を実行したり原材料を搬入したりする。その設計によっては、ドアが開かれることは稀であり、予め厳密に定められた条件下でのみ開けられる。 |
用語解説 迅速搬送システム/ポート(RTP) ーRABS又はアイソレータ内への物品搬送用のシステムで、重要区画へのリスクを最小化するもの。一例として、アルファ/ ベータのポートがある迅速搬送容器が挙げられる。 |
用語解説 エアロック ーインターロック付きドアのある囲まれた空間で、隣接する室(一般に、異なる空気清浄基準が異なるもの)間の気圧制御を保持するよう構築されたもの。エアロックの目的は、より低い管理がなされる区域から微粒子物質及び微生物の汚染が入り込むのを防ぐことである。 |
用語解説 パススルーハッチ ーエアロック(エアロックの定義を参照) と同義であるが、一般的に寸法が小さいもの。 |
パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.7 無菌製品を加工するには、陽圧管理区域を使用すること。ただし、封じ込めの理由から、病原体が露出する特定区域内の陰圧管理は許容される。特定のリスクのある原材料( 例: 病原体) の無菌処理に陰圧管理区域又はB S C を使用する場合には、適切な清浄グレードの陽圧管理ゾーンをその周囲に設けること。 |
3.10 微粒子及び微生物についての製造建屋の環境管理の度合いは、出発原料の潜在的な汚染レベル及び製品に対するリスクを考慮して、当該製品及びその製造ステップに相応したものとすること。 3.11 閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセス( 例: 添加剤、培地、緩衝液、ガスを添加する間のプロセス) では、適切な環境条件を適用すること。無菌操作については、アネックス1 に準拠したパラメータ( すなわち、グレードB を背景環境とするグレードA ) を適用すること。(略) |
3.13 閉鎖システムについて、Q R M 評価の結果に基づいて、グレードB の背景環境におけるグレードA より低い清浄度区分の区域が許容される場合があり得る。空気の清浄度区分とモニタリングの適切なレベルは、当該製品の性質、製造工程及び使用する設備を検討し、具体的なリスクを考慮して決定すること。(略) | (続き) (a) 適切な管理措置( 例:原材料及び人員の動線及び清浄度の適切な管理) を実施して微生物汚染及び交叉汚染のリスクが回避されていれば、例えば単回使用の滅菌済使い捨てキット内での加工、又はグレードC において閉鎖式の自動化製造プラットフォーム若しくは閉鎖式のフラスコ、バッグ若しくは発酵槽内での培養を用いる加工といった技術の利用が許容され得る。原材料をより高いグレードの清浄区域へ続いて移動させる場合には、特別な注意を払うこと。 |
パートA . 一般的ガイダンス 建物及び設備 5 . 管理ストラテジーの一環として、微粒子及び微生物の汚染についての製造建屋の環境管理の度合いは、出発原料の潜在的な汚染レベル及び製品に対するリスクを考慮して、当該原薬、中間製品又は最終製品及びその製造ステップに相応したものとすること。 12. 無菌製品を加工するには、陽圧管理区域を使用すること。ただし、封じ込めの理由から、病原体が露出する特定区域内の陰圧管理は許容される。特にリスクのある原材料( 例: 病原体) の無菌処理に陰圧管理区域又は安全キャビネットを使用する場合には、適切な清浄グレードの陽圧管理ゾーンを周囲に設けること。それらの気圧カスケードを明確に規定するとともに、適切なアラーム設定を行い継続的にモニターすること。 |
IV. Buildings and Facilities Clean area control parameters should be supported by microbiological and particle data obtained during qualification studies. Initial cleanroom qualification includes, in part, an assessment of air quality under as-built, static conditions. It is important for area qualification and classification to place most emphasis on data generated under dynamic conditions (i.e., with personnel present, equipment in place, and operations ongoing). An adequate aseptic processing facility monitoring program also will assess conformance with specified clean area classifications under dynamic conditions on a routine basis. The following table summarizes clean area air classifications and recommended action levels of microbiological quality (Ref. 1). |
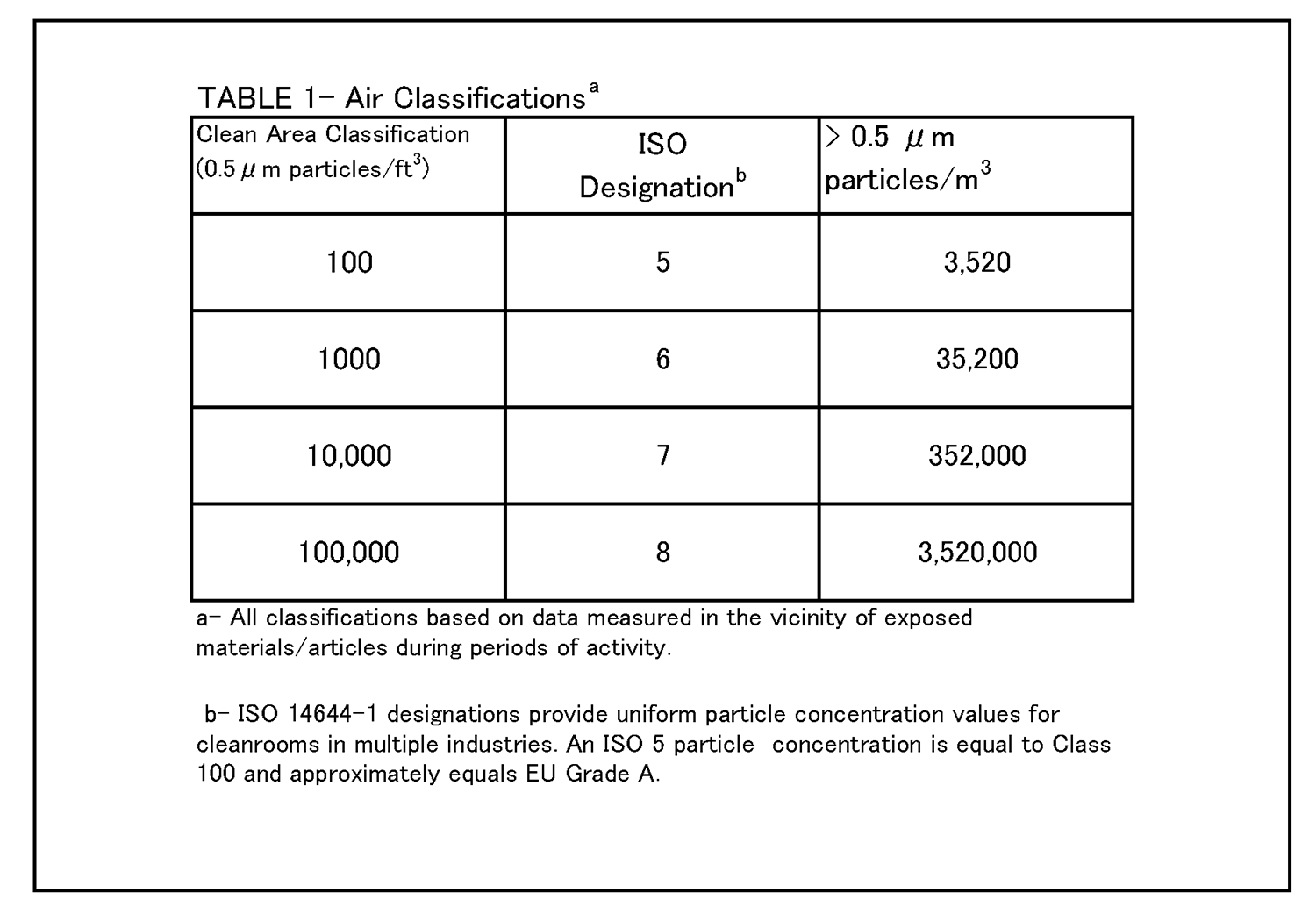 |
PREMISES AND EQUIPMENT PREMISES Production Areas 3.7. Premises should preferably be laid out in such a way as to allow the production to take place in areas connected in a logical order corresponding to the sequence of the operations and to the requisite cleanliness levels. |
4 Premises 4.4 For the manufacture of sterile products there are four grades of cleanroom/zone. Grade A: The critical zone for high-risk operations (e.g. aseptic processing line, filling zone, stopper bowl, open primary packaging or for making aseptic connections under the protection of first air). Normally, such conditions are provided by a localised airflow protection, such as unidirectional airflow workstations within RABS or isolators. The maintenance of unidirectional airflow should be demonstrated and qualified across the whole of the grade A area. |
(続き) Direct intervention (e.g. without the protection of barrier and glove port technology) into the grade A area by operators should be minimized by premises, equipment, process and procedural design. |
(続き) Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. Cleanrooms of lower grade than grade B can be considered where isolator technology is used (see paragraph 4.20). Grade C and D: These are cleanrooms used for carrying out less critical stages in the manufacture of aseptically filled sterile products or as a background for isolators. They can also be used for the preparation/filling of terminally sterilised products. (See section 8 for the specific details on terminal sterilisation activities). |
4.12 (略) Airlocks should be flushed effectively with filtered air to ensure that the grade of the cleanroom is maintained. The final stage of the airlock should, in the “at rest” state, be of the same cleanliness grade (viable and total particle) as the cleanroom into which it leads. The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (ingress/egress) by procedure should be considered.Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. Airlocks should be designed as follows: |
(続き) i.Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. ii.Material airlocks: used for materials and equipment transfer. ・Only materials and equipment that have been included on an approved list and assessed during validation of the transfer process, should be transferred into the grade A or grade B areas via an airlock or pass-through hatches. |
(続き) Equipment and materials (intended for use in the grade A area) should be protected when transiting through the grade B area. Any unapproved items that require transfer should be pre-approved as an exception. Appropriate risk assessment and mitigation measures should be applied and recorded as per the manufacturer's CCS and should include a specific disinfection and monitoring programme approved by quality assurance. |
(続き) ・Pass-through hatches should be designed to protect the higher-grade environment, for example by effective flushing with an active filtered air supply. ・The movement of material or equipment from lower grade or unclassified area to higher grade clean areas should be subject to cleaning and disinfection commensurate with the risk and in line with the CCS. |
BARRIER TECHNOLOGIES 4.20 The background environment for isolators or RABS should ensure the risk of transfer of contamination is minimized. i. Isolators: a. The background environment for open isolators should generally correspond to a minimum of grade C. The background for closed isolators should correspond to a minimum of grade D. The decision on the background classification should be based on risk assessment and justified in the CCS. ii. RABS: The background environment for RABS used for aseptic processing, should correspond to a minimum of grade B and airflow pattern studies should be performed to demonstrate the absence of air ingress during interventions, including door openings if applicable. |
Cleanroom and clean air equipment qualification 4.24 Cleanrooms and clean air equipment should be qualified using methodology in accordance with the requirements of Annex 15. Cleanroom qualification (including classification) should be clearly differentiated from operational environmental monitoring. |
4.26 Cleanroom classification is part of the cleanroom qualification and is a method of assessing the level of air cleanliness against a specification for a cleanroom or clean air equipment by measuring the total particle concentration. Classification activities should be scheduled and performed in order to avoid any impact on process or product quality. For example, initial classification should be performed during simulated operations and reclassification performed during simulated operations or during aseptic process simulation (APS). | 4.27 For cleanroom classification, the total of particles equal to or greater than 0.5 and 5 μm should be measured. This measurement should be performed both at rest and in simulated operations in accordance with the limits specified in Table 1. |
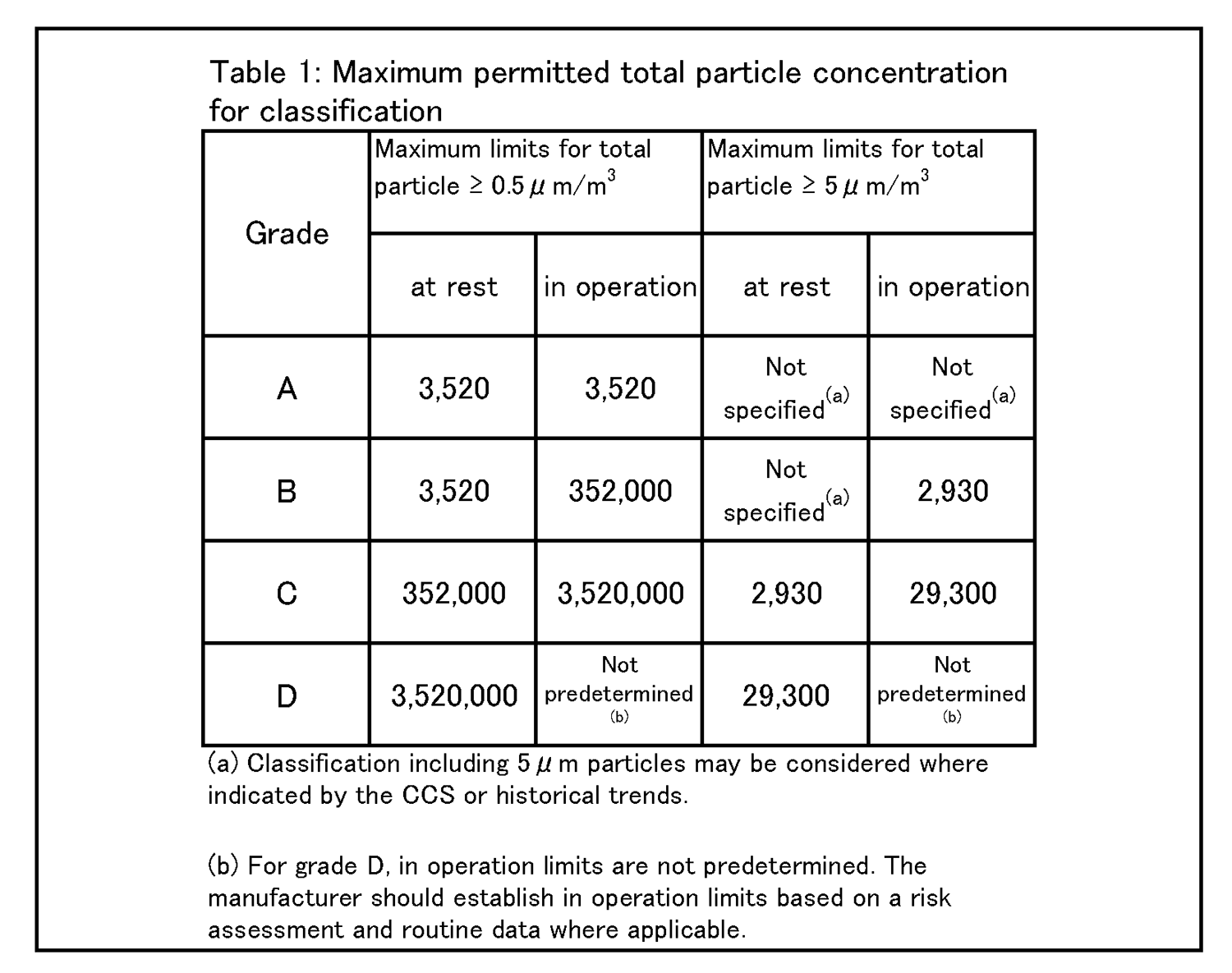 |
4.28 For classification of the cleanroom, the minimum number of sampling locations and their positioning can be found in ISO 14644 Part 1. For the aseptic processing area and the background environment (the grade A and grade B areas, respectively), additional sample locations should be considered and all critical processing areas such as the point of fill and container closure feeder bowls should be evaluated. Critical processing locations should be determined by documented risk assessment and knowledge of the process and operations to be performed in the area. | 4.29 Cleanroom classification should be carried out in the “at rest” and “in operation” states. i.The definition of “at rest” state is the condition whereby the installation of all the utilities is complete including any functioning HVAC, with the main manufacturing equipment installed as specified but not operating and without personnel present in the room. ii.The definition of “in operation” state is the condition where the installation of the cleanroom is complete, the HVAC system fully operational, equipment installed and functioning in the manufacturer’s defined operating mode with the maximum number of personnel present performing or simulating routine operational work. |
(続き) iii.The total particle limits given in Table 1 above for the “at rest” state should be achieved after a “clean up” period on completion of operations and line clearance/cleaning activities. The ""clean up"" period (guidance value of less than 20 minutes) should be determined during the qualification of the rooms, documented and adhered to in procedures to reinstate a qualified state of cleanliness if disrupted during operation. |
8 Production and Specific Technologies TERMINALLY STERILISED PRODUCTS 8.1 Preparation of components and materials should be performed in at least a grade D cleanroom in order to limit the risk of microbial, endotoxin/pyrogen and particle contamination, so that the product is suitable for sterilisation. Where the product is at a high or unusual risk of microbial contamination (e.g. the product actively supports microbial growth, the product must be held for long periods before filling or the product is not processed mostly in closed vessels), then preparation should be carried out in at least a grade C environment. |
(続き) Preparation of ointments, creams, suspensions and emulsions should be carried out in at least a grade C environment before terminal sterilisation. Specific guidance regarding terminally sterilised veterinary medicinal products can be found within Annex 4 of the GMP Guide. 8.3 Filling of products for terminal sterilisation should be carried out in at least a grade C environment. 8.4 Where the CCS identifies that the product is at an unusual risk of contamination from the environment because, for example, the filling operation is slow, the containers are wide necked or are necessarily exposed for more than a few seconds before closing, then the product should be filled in grade A with at least a grade C background. |
8.6 Examples of operations to be carried out in the various grades are given in Table 3. |
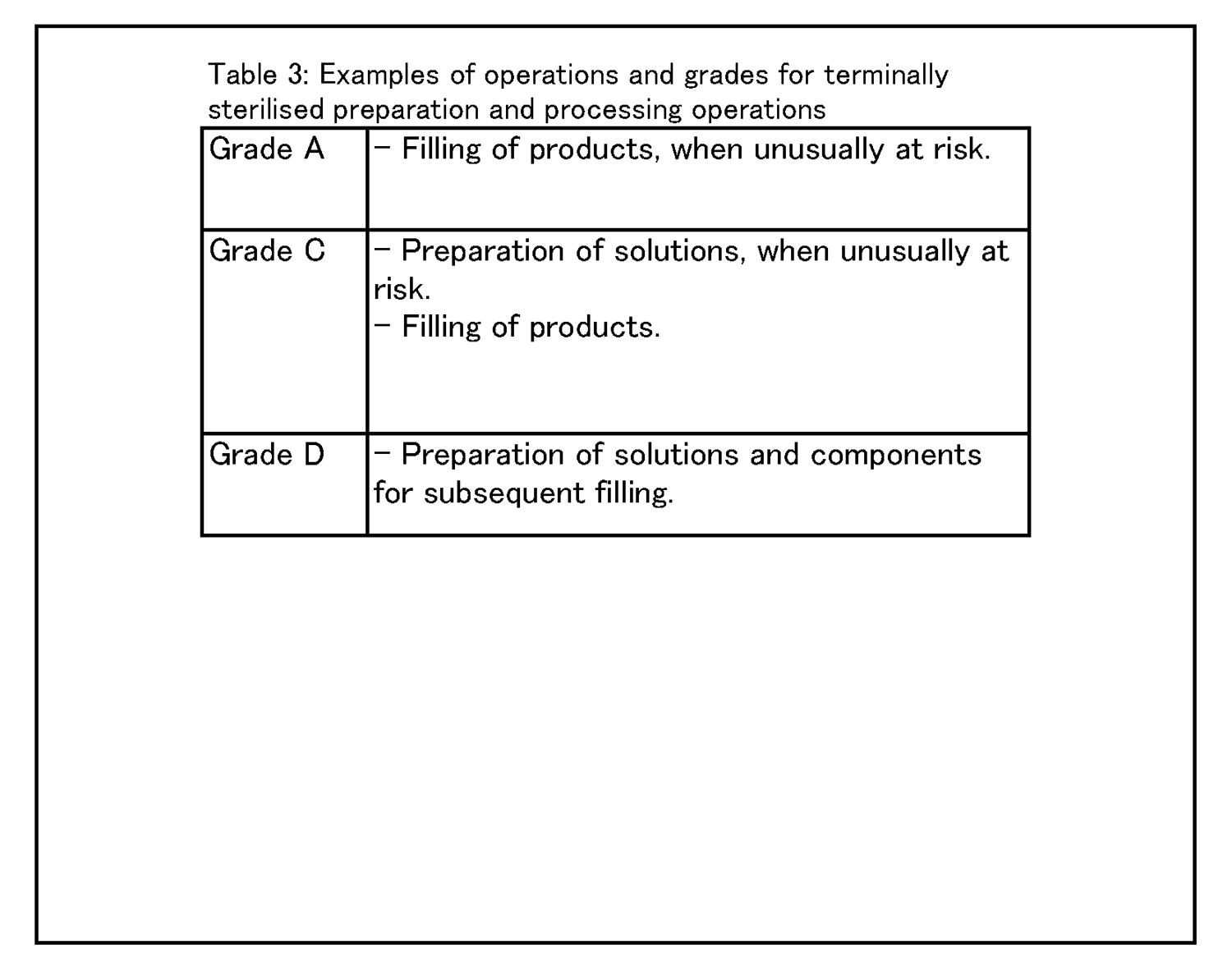 |
ASEPTIC PREPARATION AND PROCESSING 8.9 Where possible, the use of equipment such as RABS, isolators or other systems, should be considered in order to reduce the need for critical interventions into grade A and to minimize the risk of contamination. Robotics and automation of processes can also be considered to eliminate direct human critical interventions (e.g. dry heat tunnel, automated lyophilizer loading, sterilisation in place). 8.10 Examples of operations to be carried out in the various environmental grades are given in Table 4. |
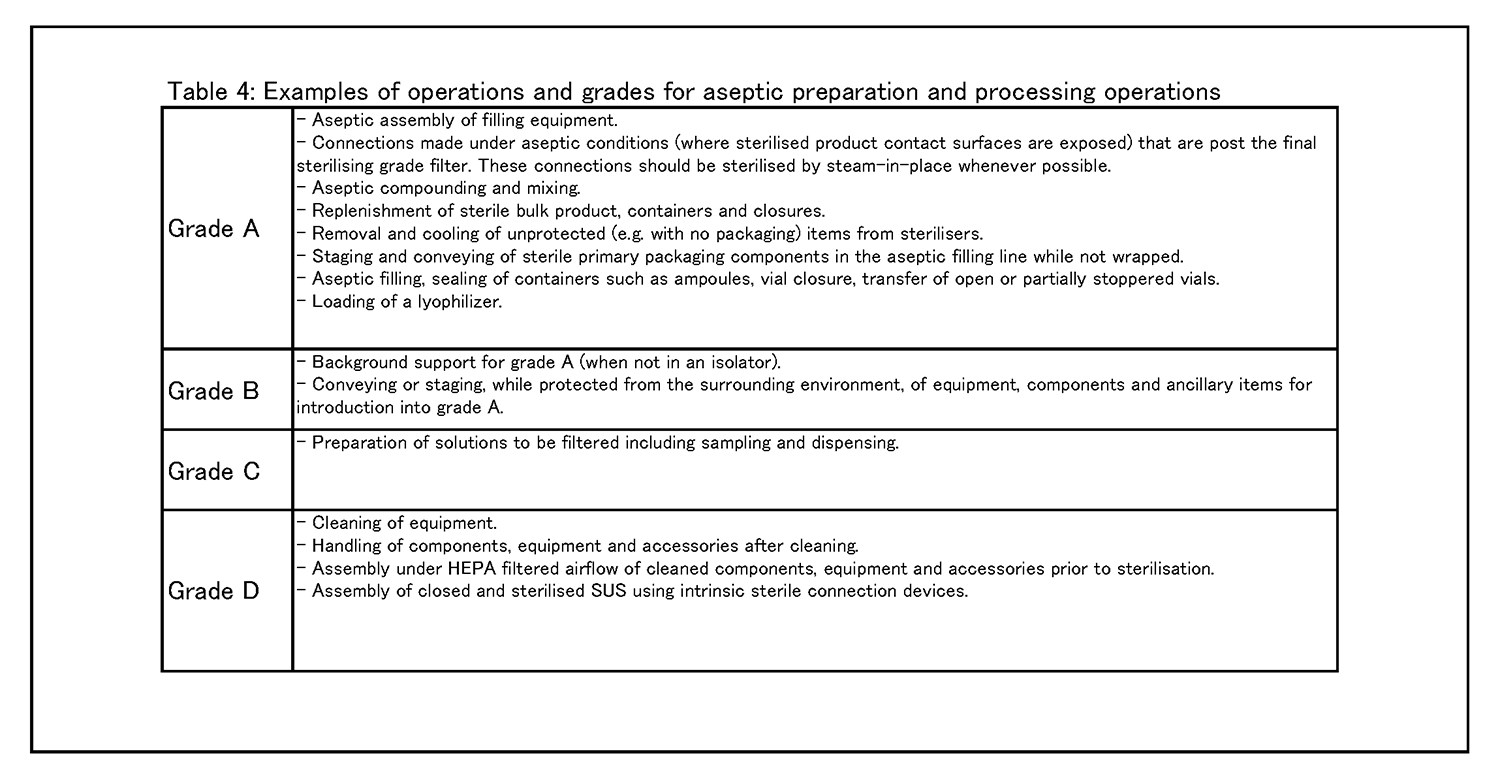 |
8.12 The unwrapping, assembly and preparation of sterilised equipment, components and ancillary items with direct or indirect product contact should be treated as an aseptic process and performed in grade A with a grade B background. The filling line set-up and filling of the sterile product should be treated as an aseptic process and performed in grade A with a grade B background. Where an isolator is used, the background should be in accordance with paragraph 4.20. | 8.13 Preparation and filling of sterile products such as ointments, creams, suspensions and emulsions should be performed in grade A with a grade B background when the product and components are exposed to the environment and the product is not subsequently filtered (via a sterilising grade filter) or terminally sterilised. Where an isolator or RABS is used, the background should be in accordance with paragraph 4.20. | 8.14 Aseptic connections should be performed in grade A with a grade B background unless subsequently sterilised in place or conducted with intrinsic sterile connection devices that minimize any potential contamination from the immediate environment. Intrinsic sterile connection devices should be designed to mitigate risk of contamination. Where an isolator is used, the background should be in accordance with paragraph 4.20. Aseptic connections should be appropriately assessed and their effectiveness verified. For requirements regarding ntrinsic sterile connection devices, see paragraphs 8.129 and 8.130. |
9 Environmental & process monitoring 9.9 Appropriate alert levels and action limits should be set for the results of viable and total particle monitoring. The maximum total particle action limits are described in Table 5 and the maximum viable particle action limits are described in Table 6. |
9.12 The monitoring of grade C and D cleanrooms in operation should be performed based on data collected during qualification and routine data to allow effective trend analysis. The requirements of alert levels and action limits will depend on the nature of the operations carried out. Action limits may be more stringent than those listed in Table 5 and Table 6. Environmental monitoring - Total Particle 9.15 The limits for environmental monitoring of airborne particle concentration for each graded area are given in Table 5. |
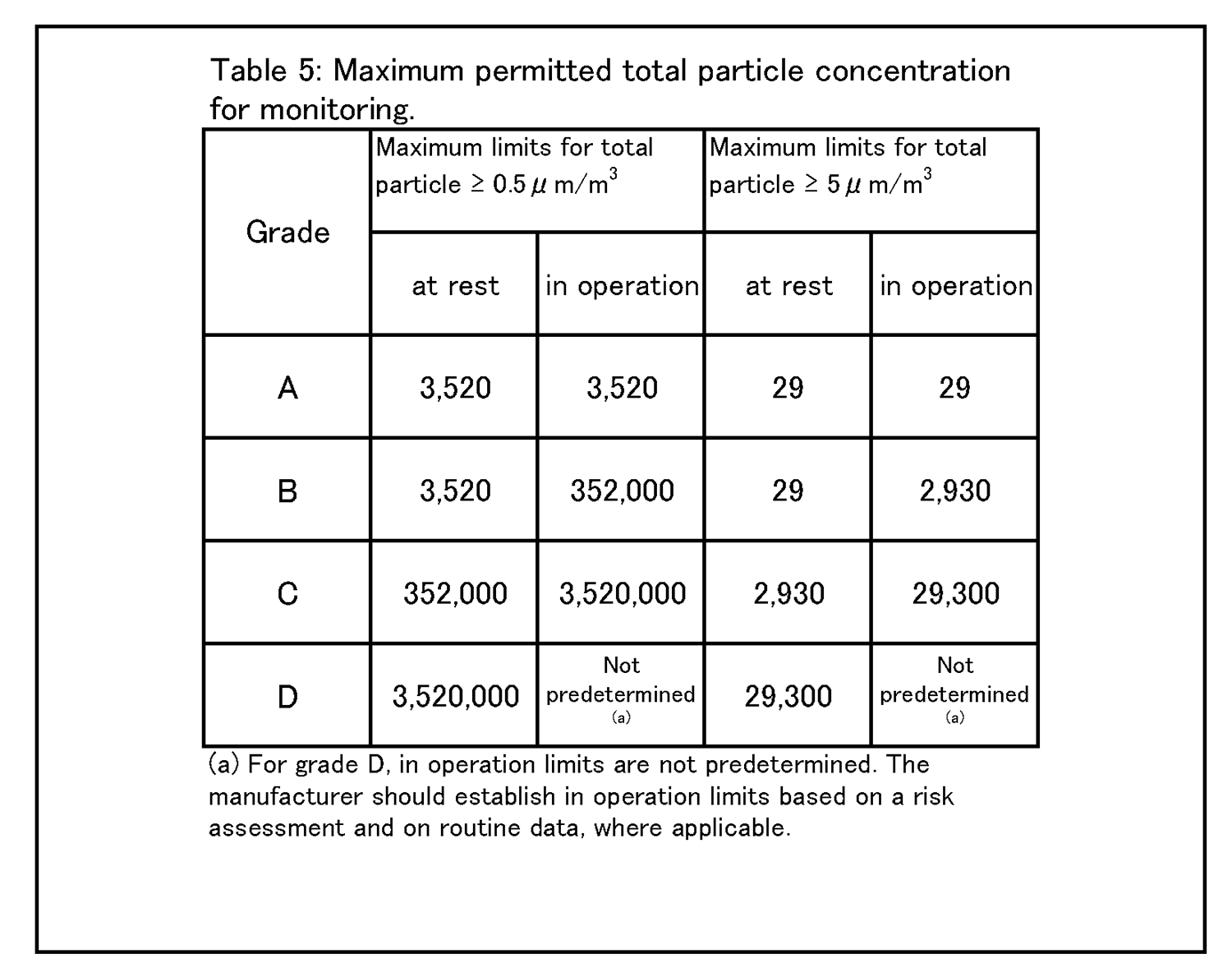 |
Note 1:The particle limits given in the table for the “at rest” state should be achieved after a short “clean up” period defined during qualification (guidance value of less than 20 minutes) in an unmanned state, after the completion of operations (see paragraph 4.29). Note 2:The occasional indication of macro particle counts, especially ≧ 5 μm, within grade A may be considered to be false counts due to electronic noise, stray light, coincidence loss etc. However, consecutive or regular counting of low levels may be indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the room air supply filtration system, equipment failure, or may also be diagnostic of poor practices during machine set-up and routine operation. |
Glossary Isolator - An enclosure capable of being subject to reproducible interior bio-decontamination, with an internal work zone meeting grade A conditions that provides uncompromised, continuous isolation of its interior from the external environment (e.g. surrounding cleanroom air and personnel). There are two major types of isolators: i. Closed isolator systems exclude external contamination of the isolator’s interior by accomplishing material transfer via aseptic connection to auxiliary equipment, rather than use of openings to the surrounding environment. Closed systems remain sealed throughout operations. |
(続き) ii. Open isolator systems are designed to allow for the continuous or semi-continuous ingress and/or egress of materials during operations through one or more openings. Openings are engineered (e.g. using continuous overpressure) to exclude the entry of external contaminant into the isolator. |
Glossary Restricted Access Barrier System (RABS) - System that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A), and using a rigid-wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, RTPs and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened, and only under strictly pre-defined conditions. |
Glossary Rapid Transfer System/Port (RTP) - A System used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
PART A: GENERAL GUIDANCE SUPPLIMENTARY PROVISIONS TO PIC/S GMP GUIDE PART I CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Production Areas 3.7 Positive pressure areas should be used to process sterile products, but negative pressure in specific areas at the point of exposure of pathogens is acceptable for containment reasons. Where negative pressure areas or BSCs are used for aseptic processing of materials with particular risks (e.g. pathogens), they should be surrounded by a positive pressure clean zone of appropriate Grade. |
3.10 The degree of environmental control of particulate and microbial contamination of the production premises should be adapted to the product and the production step, bearing in mind the potential level of contamination of the starting materials and the risks to the product. 3.11 Where processes are not closed and there is exposure of the product to the immediate room environment without a subsequent microbial inactivation process, (e.g. during additions of supplements, media, buffers, gasses, manipulations) appropriate environmental conditions should be applied. For aseptic manipulations parameters in line with Annex 1 (i.e. Grade A with Grade B background) should be applied. (略) |
3.13 For closed systems, a lower classified area than Grade A in background Grade B might be acceptable based on the outcome of a QRM assessment. The appropriate level of air classification and monitoring should be determined having regard to the specific risks, considering the nature of the product, the manufacturing process and the equipment used. QRM should be used to determine whether the technology used supports reduced monitoring, in particular where monitoring can be a source of contamination. This is in addition to: | (続き) (a) The use of technologies as e.g. processing inside single use sterile disposable kits, or processing using closed, automated manufacturing platform or incubation in closed flasks, bags or fermenters in Grade C may be acceptable if adequate control measures are implemented to avoid the risk of microbial contamination and cross-contamination (e.g. appropriate control of materials, personnel flows and cleanliness). Particular attention should be paid if the materials are subsequently moved to a clean area of higher Grade. |
Chapter 3: Premises and Equipment Production Area 3.7 Premises should preferably be laid out in such a way as to allow the production to take place in areas connected in a logical order corresponding to the sequence of the operations and to the requisite cleanliness levels. |
4.4 For the manufacture of sterile products, there are four grades of cleanroom/zone. Grade A: The critical zone for high-risk operations (e.g. aseptic processing line, filling zone, stopper bowl, open primary packaging or for making aseptic connections under the protection of first air). Normally, such conditions are provided by a localised airflow protection, such as unidirectional airflow workstations within RABS or isolators. The maintenance of unidirectional airflow should be demonstrated and qualified across the whole of the grade A area. |
(続き) Direct intervention (e.g. without the protection of barrier and glove port technology) into the grade A area by operators should be minimized by premises, equipment, process and procedural design. |
(続き) Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. Cleanrooms of lower grade than grade B can be considered where isolator technology is used (see paragraph 4.20 ). Grade C and D: These are cleanrooms used for carrying out less critical stages in the manufacture of aseptically filled sterile products or as a background for isolators. They can also be used for the preparation/filling of terminally sterilised products. (See section 8 for the specific details on terminal sterilisation activities). |
4.12 (略) Airlocks should be flushed effectively with filtered air to ensure that the grade of the cleanroom is maintained. The final stage of the airlock should, in the “at rest” state, be of the same cleanliness grade (viable and total particle) as the cleanroom into which it leads. The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (ingress/egress) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. Airlocks should be designed as follows: |
(続き) i.Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. ii.Material airlocks: used for materials and equipment transfer. ・Only materials and equipment that have been included on an approved list and assessed during validation of the transfer process should be transferred into the grade A or grade B areas via an airlock or pass-through hatches. |
(続き) Equipment and materials (intended for use in the grade A area) should be protected when transiting through the grade B area. Any unapproved items that require transfer should be pre-approved as an exception. Appropriate risk assessment and mitigation measures should be applied and recorded as per the manufacturer's CCS and should include a specific disinfection and monitoring programme approved by quality assurance. |
(続き) ・Pass-through hatches should be designed to protect the higher-grade environment, for example by effective flushing with an active filtered air supply ・The movement of material or equipment from lower grade or unclassified area to higher-grade clean areas should be subject to cleaning and disinfection commensurate with the risk and in line with the CCS. |
Barrier Technologies 4.20 The background environment for isolators or RABS should ensure the risk of transfer of contamination is minimized. i. Isolators: a. The background environment for open isolators should generally correspond to a minimum of grade C. The background for closed isolators should correspond to a minimum of grade D. The decision on the background classification should be based on risk assessment and justified in the CCS. ii. RABS: The background environment for RABS used for aseptic processing should correspond to a minimum of grade B and airflow pattern studies should be performed to demonstrate the absence of air ingress during interventions, including door openings if applicable. |
Cleanroom and clean air equipment qualification 4.24 Cleanrooms and clean air equipment should be qualified using methodology in accordance with the requirements of Annex 15. Cleanroom qualification (including classification) should be clearly differentiated from operational environmental monitoring. |
4.26 Cleanroom classification is part of the cleanroom qualification and is a method of assessing the level of air cleanliness against a specification for a cleanroom or clean air equipment by measuring the total particle concentration. Classification activities should be scheduled and performed in order to avoid any impact on process or product quality. For example, initial classification should be performed during simulated operations and reclassification performed during simulated operations or during aseptic process simulation (APS). | 4.27 For cleanroom classification, the total of particles equal to or greater than 0.5 and 5 μm should be measured. This measurement should be performed both at rest and in simulated operations in accordance with the limits specified in Table 1. |
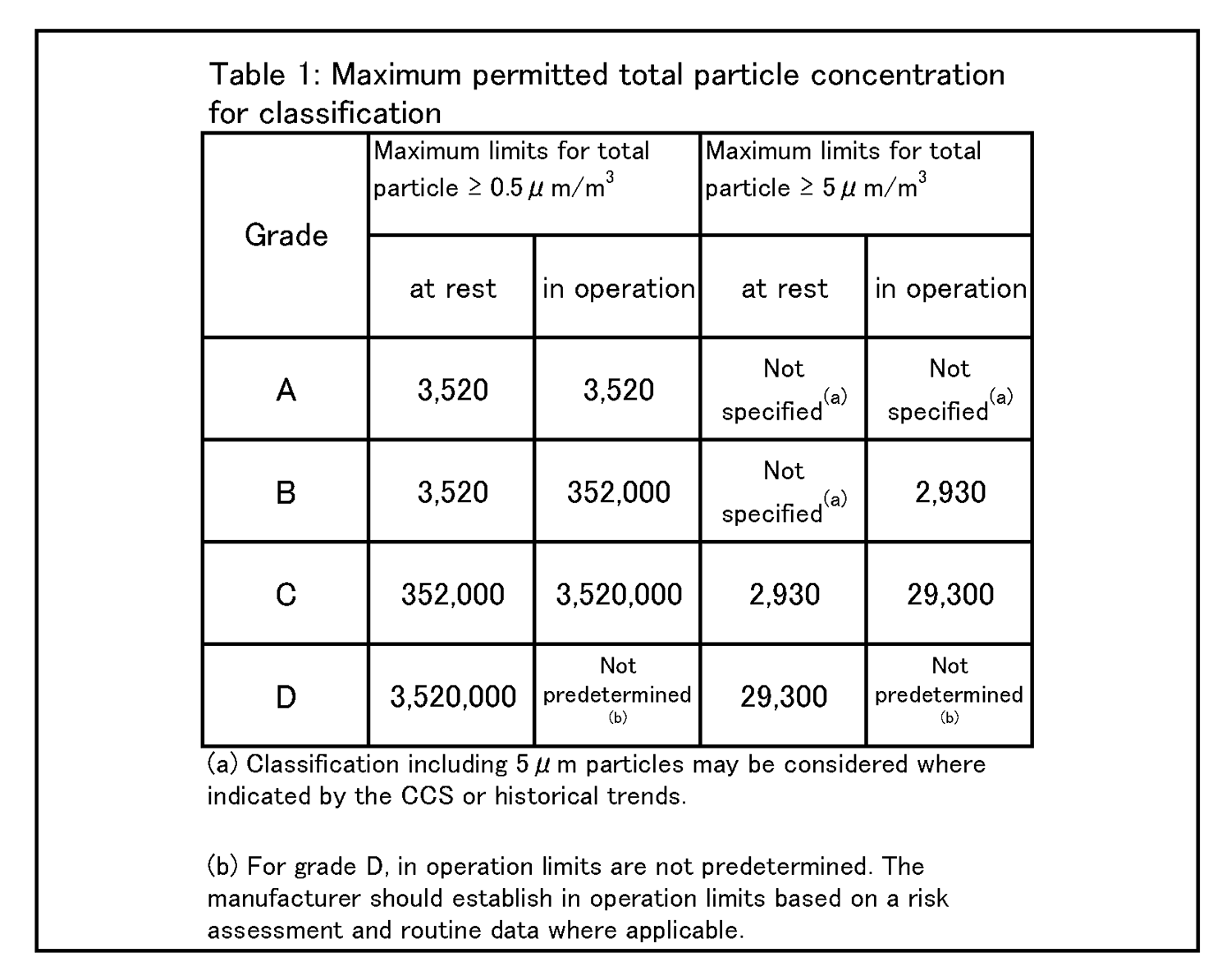 |
4.28 For classification of the cleanroom, the minimum number of sampling locations and their positioning can be found in ISO 14644 Part 1. For the aseptic processing area and the background environment (the grade A and grade B areas, respectively), additional sample locations should be considered and all critical processing areas such as the point of fill and container closure feeder bowls should be evaluated. Critical processing locations should be determined by documented risk assessment and knowledge of the process and operations to be performed in the area. | 4.29 Cleanroom classification should be carried out in the “at rest” and “in operation” states. i.The definition of “at rest” state is the condition whereby the installation of all the utilities is complete including any functioning HVAC, with the main manufacturing equipment installed as specified but not operating and without personnel present in the room. ii.The definition of “in operation” state is the condition where the installation of the cleanroom is complete, the HVAC system fully operational, equipment installed and functioning in the manufacturer’s defined operating mode with the maximum number of personnel present performing or simulating routine operational work. |
(続き) iii.The total particle limits given in Table 1 above for the “at rest” state should be achieved after a “clean up” period on completion of operations and line clearance/cleaning activities. The ""clean up"" period (guidance value of less than 20 minutes) should be determined during the qualification of the rooms, documented and adhered to in procedures to reinstate a qualified state of cleanliness if disrupted during operation. |
8 Production and Specific Technologies Terminally sterilised products 8.1 Preparation of components and materials should be performed in at least a grade D cleanroom in order to limit the risk of microbial, endotoxin/pyrogen and particle contamination, so that the product is suitable for sterilisation. Where the product is at a high or unusual risk of microbial contamination (e.g. the product actively supports microbial growth, the product must be held for long periods before filling or the product is not processed mostly in closed vessels), then preparation should be carried out in at least a grade C environment. |
(続き) Preparation of ointments, creams, suspensions and emulsions should be carried out in at least a grade C environment before terminal sterilisation. Specific guidance regarding terminally sterilised veterinary medicinal products can be found within Annex 4 of the GMP guidelines. 8.3 Filling of products for terminal sterilisation should be carried out in at least a grade C environment. 8.4 Where the CCS identifies that the product is at an unusual risk of contamination from the environment because, for example, the filling operation is slow, the containers are wide necked or are necessarily exposed for more than a few seconds before closing, then the product should be filled in grade A with at least a grade C background. |
8.6 Examples of operations to be carried out in the various grades are given in Table 3. |
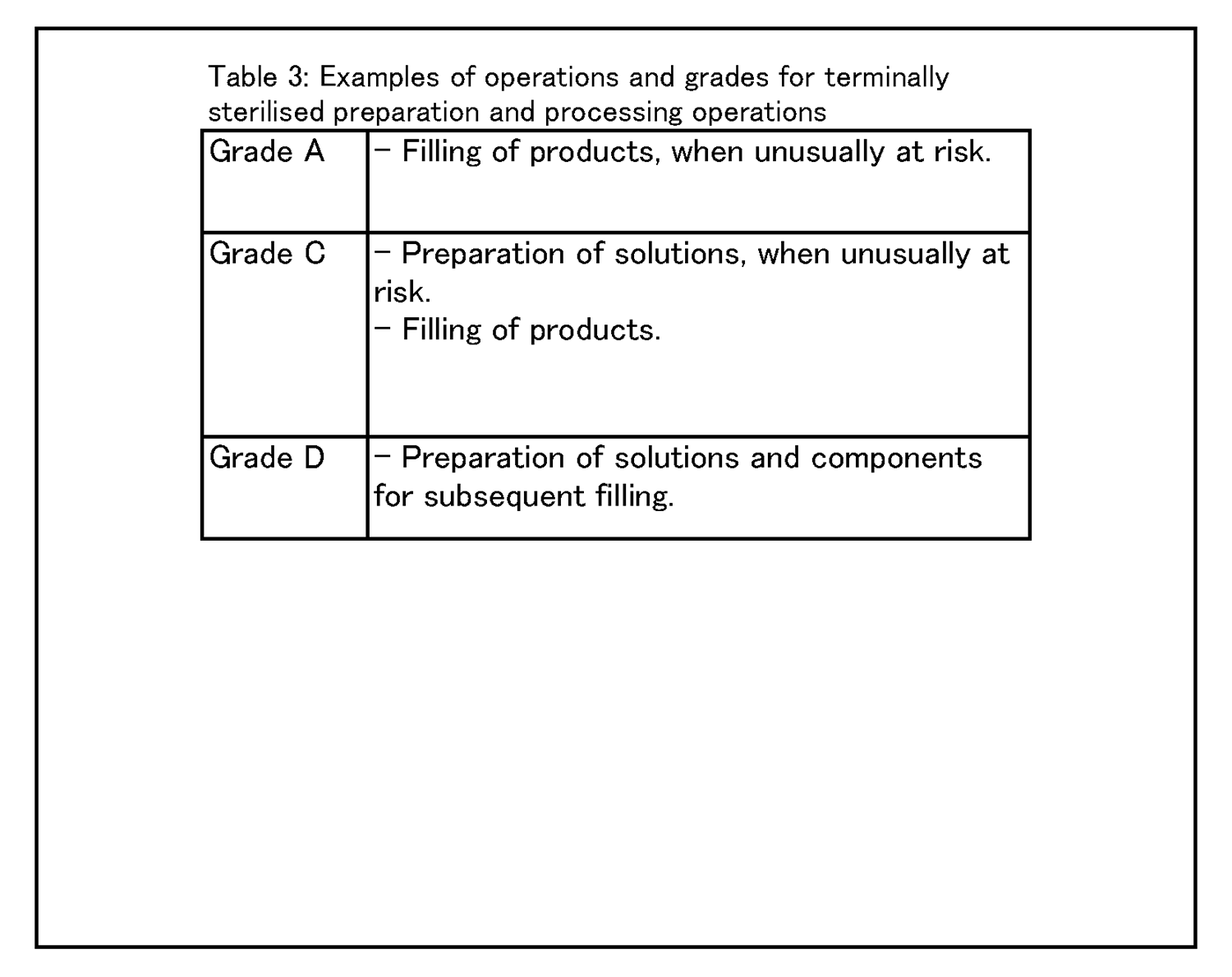 |
Aseptic preparation and processing 8.9 Where possible, the use of equipment such as RABS, isolators or other systems, should be considered in order to reduce the need for critical interventions into grade A and to minimize the risk of contamination. Robotics and automation of processes can also be considered to eliminate direct human critical interventions (e.g. dry heat tunnel, automated lyophilizer loading, sterilisation in place). 8.10 Examples of operations to be carried out in the various environmental grades are given in Table 4. |
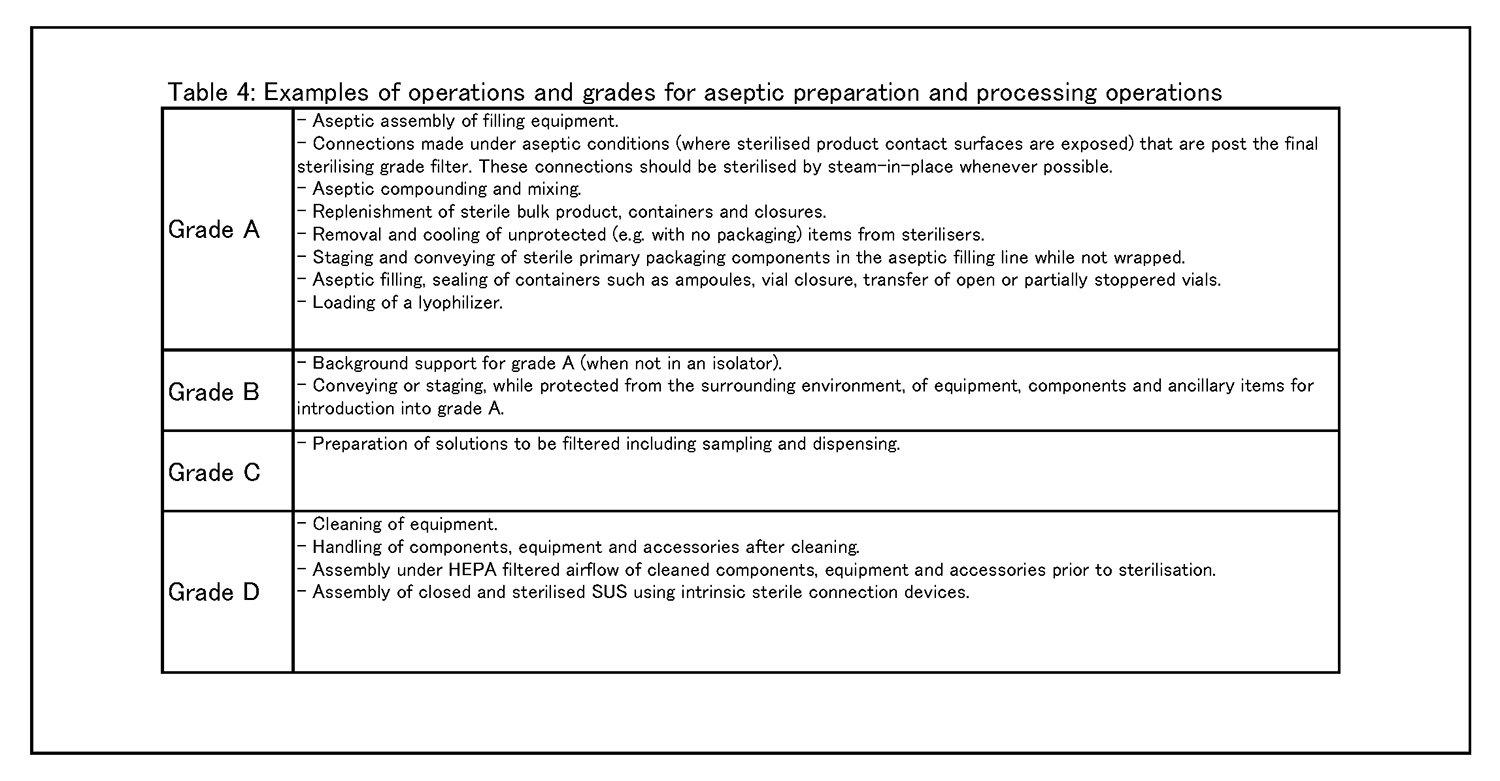 |
8.12 The unwrapping, assembly and preparation of sterilised equipment, components and ancillary items with direct or indirect product contact should be treated as an aseptic process and performed in grade A with a grade B background. The filling line set-up and filling of the sterile product should be treated as an aseptic process and performed in grade A with a grade B background. Where an isolator is used, the background should be in accordance with paragraph 4.20. | 8.13 Preparation and filling of sterile products such as ointments, creams, suspensions and emulsions should be performed in grade A with a grade B background when the product and components are exposed to the environment and the product is not subsequently filtered (via a sterilising grade filter) or terminally sterilised. Where an isolator or RABS is used, the background should be in accordance with paragraph 4.20. | 8.14 Aseptic connections should be performed in grade A with a grade B background unless subsequently sterilised in place or conducted with intrinsic sterile connection devices that minimize any potential contamination from the immediate environment. Intrinsic sterile connection devices should be designed to mitigate risk of contamination. Where an isolator is used, the background should be in accordance with paragraph 4.20. Aseptic connections should be appropriately assessed and their effectiveness verified. For requirements regarding intrinsic sterile connection devices see paragraphs 8.129 and 8.130. |
9 Environmental & process monitoring Environmental monitoring - total particle 9.9 Appropriate alert levels and action limits should be set for the results of viable and total particle monitoring. The maximum total particle action limits are described in Table 5 and the maximum viable particle action limits are described in Table 6. |
9.12 The monitoring of grade C and D cleanrooms in operation should be performed based on data collected during qualification and routine data to allow effective trend analysis. The requirements of alert levels and action limits will depend on the nature of the operations carried out. Action limits may be more stringent than those listed in Table 5 and Table 6. Environmental monitoring - Total Particle 9.15 The limits for environmental monitoring of airborne particle concentration for each graded area are given in Table 5. |
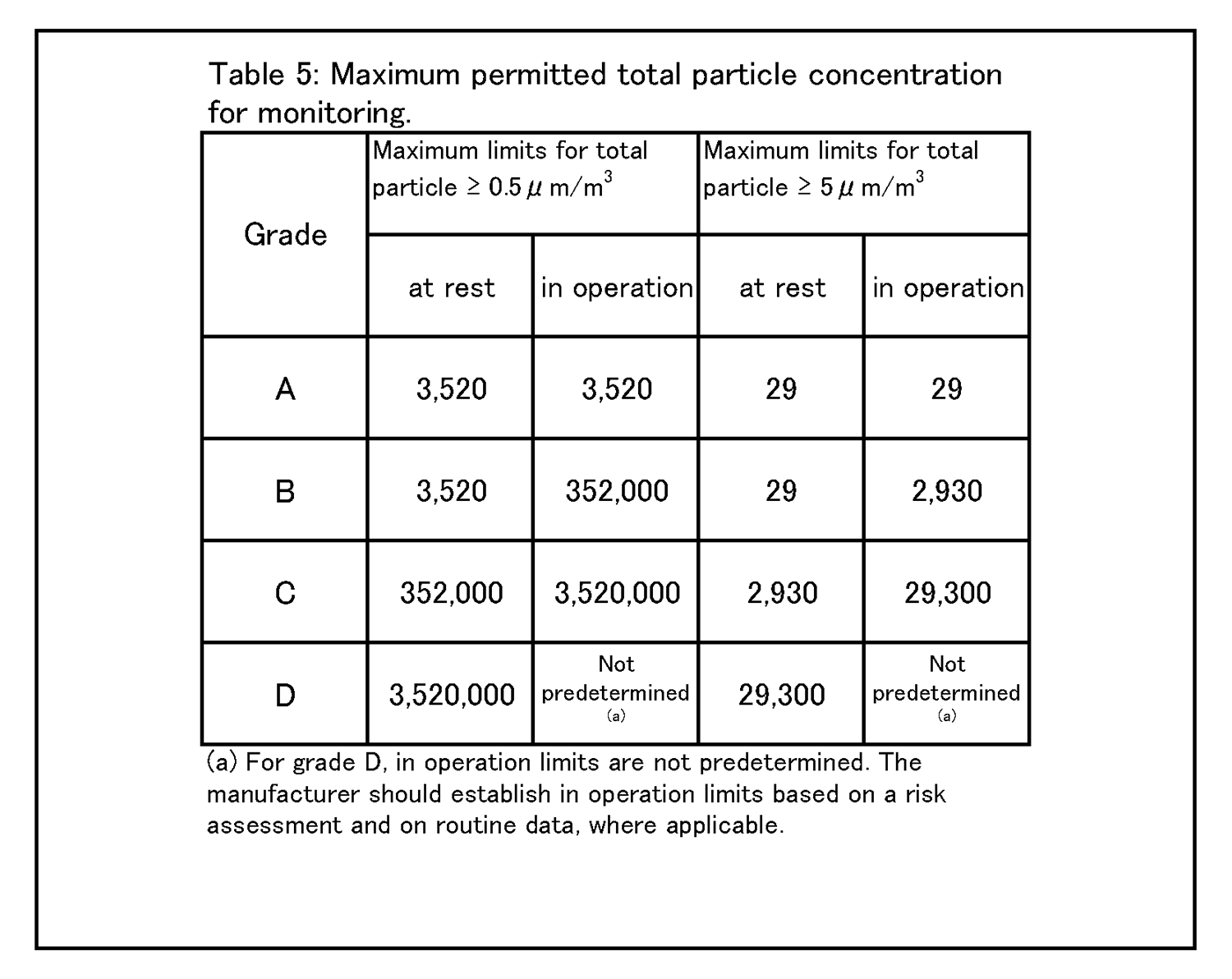 |
Note 1: The particle limits given in the table for the “at rest” state should be achieved after a short “clean up” period defined during qualification (guidance value of less than 20 minutes) in an unmanned state, after the completion of operations (see paragraph 4.29). Note 2: The occasional indication of macro particle counts, especially ≧ 5 μm, within grade A may be considered to be false counts due to electronic noise, stray light, coincidence loss etc. However, consecutive or regular counting of low levels may be indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the room air supply filtration system, equipment failure, or may also be diagnostic of poor practices during machine set-up and routine operation. |
Glossary Isolator - An enclosure capable of being subject to reproducible interior bio-decontamination, with an internal work zone meeting grade A conditions that provides uncompromised, continuous isolation of its interior from the external environment (e.g. surrounding cleanroom air and personnel). There are two major types of isolators: i. Closed isolator systems exclude external contamination of the isolator’s interior by accomplishing material transfer via aseptic connection to auxiliary equipment, rather than use of openings to the surrounding environment. Closed systems remain sealed throughout operations. |
(続き) ii. Open isolator systems are designed to allow for the continuous or semi-continuous ingress and/or egress of materials during operations through one or more openings. Openings are engineered (e.g. using continuous overpressure) to exclude the entry of external contaminant into the isolator. |
Glossary Restricted Access Barrier System (RABS) - System that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A), and using a rigid-wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, RTPs and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened, and only under strictly pre-defined conditions. |
Glossary Rapid Transfer System/Port (RTP) - A System used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
4 Premises 4.4 Four grades of cleanrooms or zones are normally used for the manufacture of sterile products. Grade A. This is the critical zone for high-risk operations (for example, aseptic processing line, filling zone, stopper bowl, open primary packaging, or for making aseptic connections under the protection of first air). Normally, such conditions are provided by a localized airflow protection, such as unidirectional airflow work stations within RABS or isolators. The maintenance of unidirectional airflow should be demonstrated and qualified across the whole of the grade A area. Direct intervention (for example, without the protection of barrier and glove port technology) into the grade A area by operators should be minimized by premises, equipment, process and procedural design. |
(続き) Grade B. For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Where applicable, air pressure differential between grade B and an adjacent area should be continuously monitored. Cleanrooms of lower grade than grade B can be considered where isolator technology is used (refer to paragraph 4.20). Grades C and D. These are cleanrooms used for carrying out less critical stages in the manufacture of aseptically filled sterile products or as a background for isolators. They can also be used for the preparation or filling of terminally sterilized products (see section 8 for specific details on terminal sterilization activities). |
4.12 (略) Airlocks should be effectively flushed with filtered air to ensure that the grade of the cleanroom is maintained. The final airlock should, in the at rest state, be of the same cleanliness grade (viable and total particle) as the cleanroom into which it leads. The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (inward or outward) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. Airlocks should be designed as follows: |
(続き) i.Personnel airlocks: areas of increasing cleanliness used for entry of personnel (for example, from the grade D area to the grade C area to the grade B area). In general, handwashing facilities should be provided only in the first change room and should not be present in change rooms directly accessing the grade B area. ii.Material airlocks: used for materials and equipment transfer. - Only materials and equipment that have been included on an approved list and assessed during validation of the transfer process should be transferred into the grade A or B areas via an airlock or pass-through hatch. |
(続き) Equipment and materials intended for use in the grade A area should be protected when transiting through the grade B area. Any unapproved items that require transfer should be preapproved as an exception. Appropriate risk assessment and mitigation measures should be applied and recorded as per the manufacturer’s CCS and should include a specific disinfection and monitoring programme approved by quality assurance. |
(続き) - Pass-through hatches should be designed to protect the highergrade environment, for example by effective flushing with active filtered air supply of appropriate grade in accordance with the CCS. - The movement of material or equipment from lower-grade or unclassified areas to higher-grade clean areas should be subject to cleaning and disinfection commensurate with the risk and in line with the CCS. |
Barrier technologies 4.20 The background environment for isolators and RABS should ensure that the risk of transfer of contamination is minimized. i. Isolators: a. The background environment for open isolators should generally correspond to a minimum of grade C. The background for closed isolators should correspond to a minimum of grade D. The decision on the background classification should be based on risk assessment and justified in the CCS. ii. RABS: a. The background environment for RABS used for aseptic processing should correspond to a minimum of grade B, and airflow pattern studies should be performed to demonstrate the absence of air ingress during interventions, including door openings if applicable. |
Cleanroom and clean air equipment 4.24 Cleanrooms and clean air equipment should be qualified using methodology in accordance with the requirements of the WHO Good manufacturing practices: guideline on validation.3 Cleanroom qualification (including classification) should be clearly differentiated from operational environmental monitoring. |
4.26 Cleanroom classification is part of the cleanroom qualification and is a method of confirming the level of air cleanliness against a specification for a cleanroom or clean air equipment by measuring the particle concentration. Classification activities should be scheduled and performed in order to avoid any impact on process or product quality. For example,initial classification should be performed during simulated operations and reclassification performed during simulated operations or during aseptic process simulation (APS). | 4.27 For cleanroom classification, the total of particles equal to or greater than 0.5 and 5 μm should be measured. Maximum permitted particle concentration limits are specified in Table. 1. |
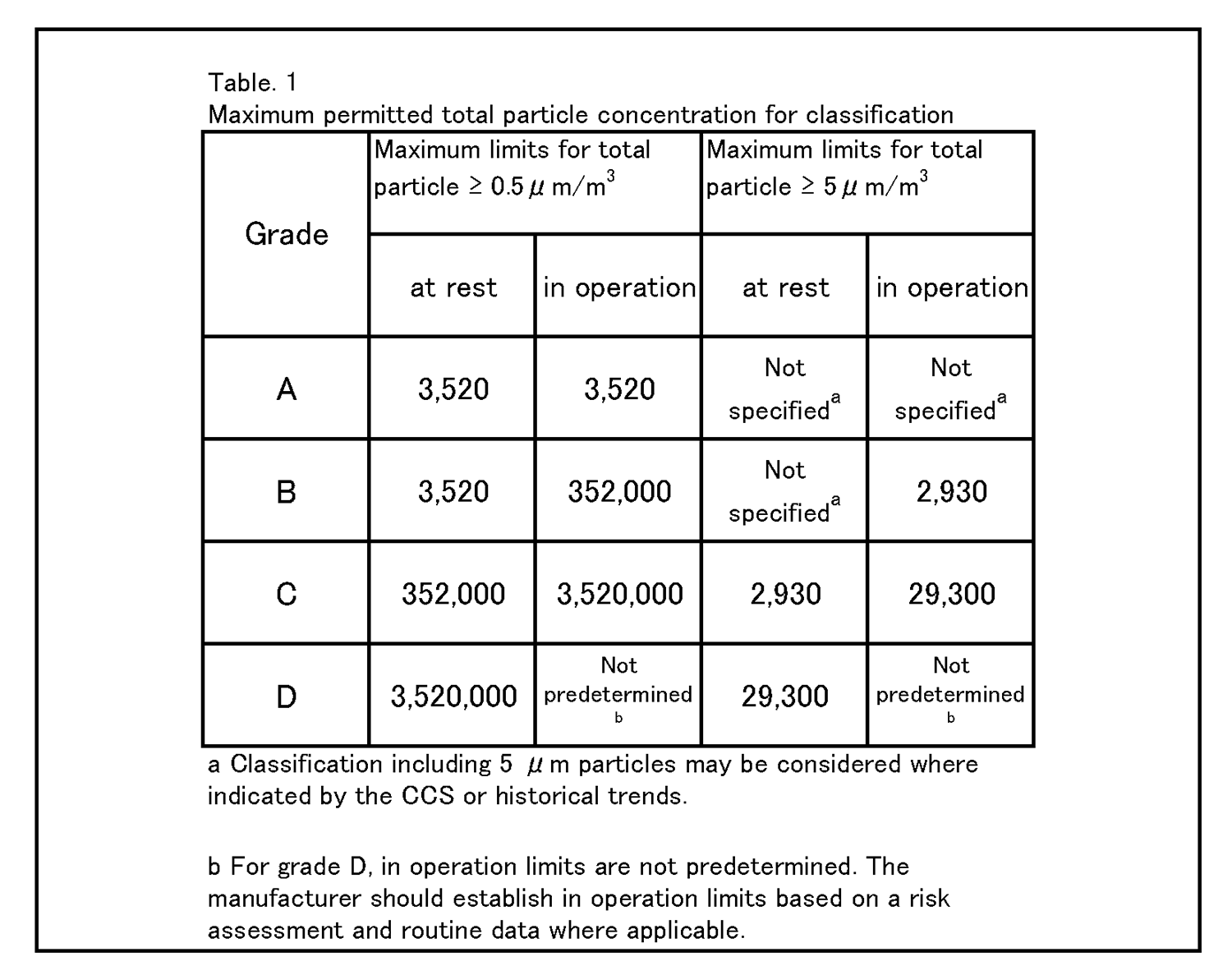 |
4.28 For classification of the cleanroom, the minimum number of sampling locations and their positioning can be found in ISO 14644 Part 1. For the aseptic processing area and the background environment (the grade A and B areas, respectively) additional sample locations should be considered, and all critical processing areas, such as the point of fill and container closure feeder bowls, should be evaluated. Critical processing locations should be determined by documented risk assessment and knowledge of the process and operations to be performed in the area. | 4.29 Cleanroom classification should be carried out in the at rest and in operation states. i.The definition of the at rest state is the condition whereby the installation of all the utilities is complete, including any functioning HVAC, with the main manufacturing equipment installed as specified but not operating and without personnel present in the room. ii.The definition of the in operation state is the condition whereby the installation of the cleanroom is complete, the HVAC system fully operational, and the equipment is installed and functioning in the manufacturer’s defined operating mode, with the maximum number of personnel present performing or simulating routine operational work. |
(続き) iii.The total particle limits given in Table.1 above for the at rest state should be achieved after a clean-up period upon completion of operations and line clearance or cleaning activities. The clean-up period (guidance value of less than 20 minutes) should be determined during the qualification of the rooms, documented, and adhered to in procedures to reinstate a qualified state of cleanliness if disrupted during operation. |
8 Production and Specific Technologies Terminally sterilized products 8.1 Preparation of components and materials should be performed in at least a grade D cleanroom in order to limit the risk of microbial, endotoxin/pyrogen and particle contamination, so that the product is suitable for sterilization. Where the product is at a high or unusual risk of microbial contamination (for example, the product actively supports microbial growth and must be held for long periods before filling, or the product is not processed mostly in closed vessels), then preparation should be carried out in at least a grade C environment. |
(続き) The preparation of ointments, creams, suspensions and emulsions should be carried out in at least a grade C environment before terminal sterilization. 8.3 The filling of products for terminal sterilization should be carried out in at least a grade C environment. 8.4 Where the CCS identifies that the product is at an unusual risk of contamination from the environment - for example, when the filling operation is slow or when the containers are wide necked or are necessarily exposed for more than a few seconds before closing - then the product should be filled in grade A with at least a grade C background. |
8.6 Examples of operations to be carried out in the various grades are given in Table. 3. |
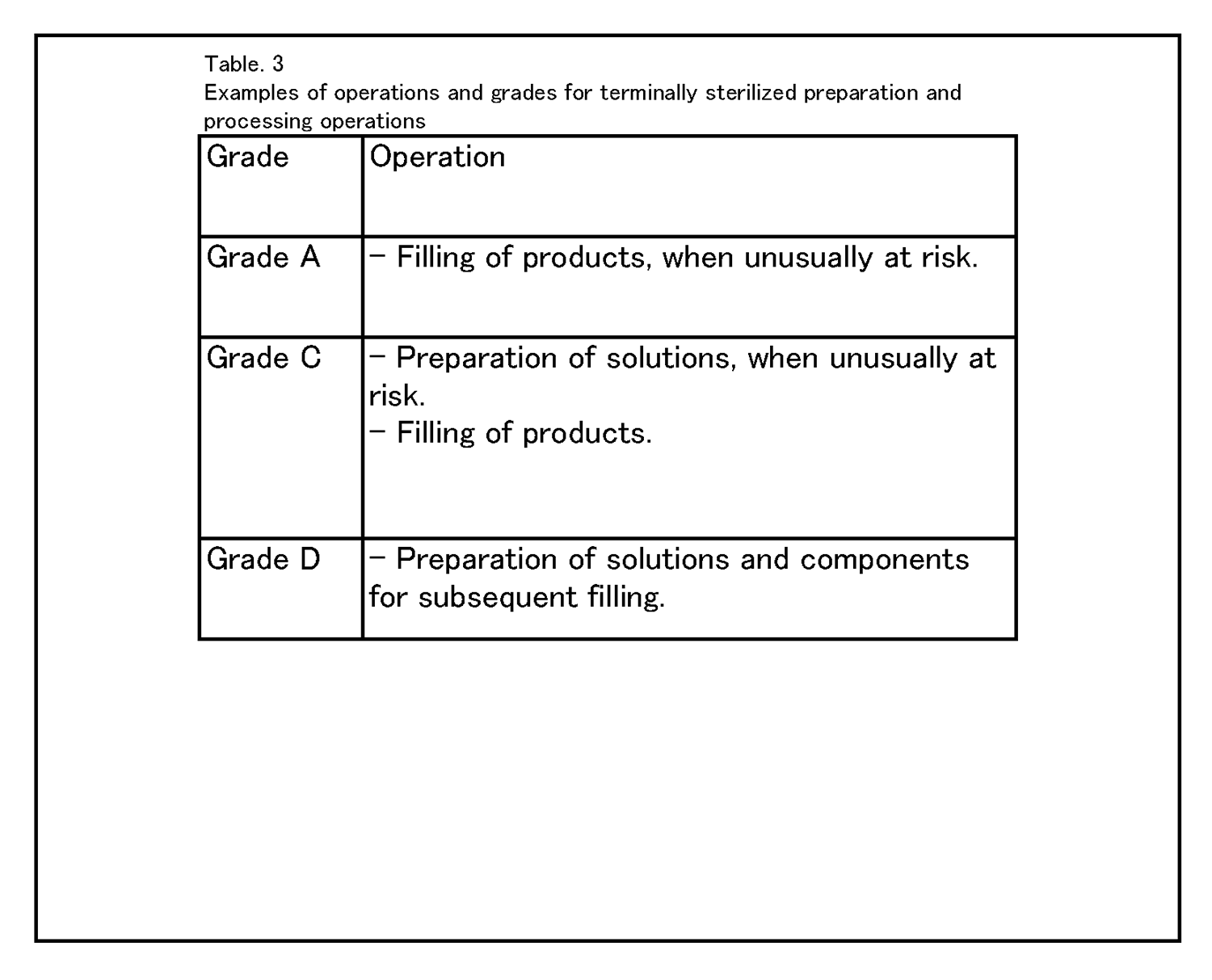 |
Aseptic preparation and processing 8.9 Where possible, the use of equipment such as RABS, isolators or other systems should be considered in order to reduce the need for critical interventions into grade A and to minimize the risk of contamination. Robotics and automation of processes can also be considered to eliminate direct human critical interventions (for example, dry heat tunnel, automated lyophilizer loading, sterilization in place). 8.10 Examples of operations to be carried out in the various environmental grades are given in Table 4. |
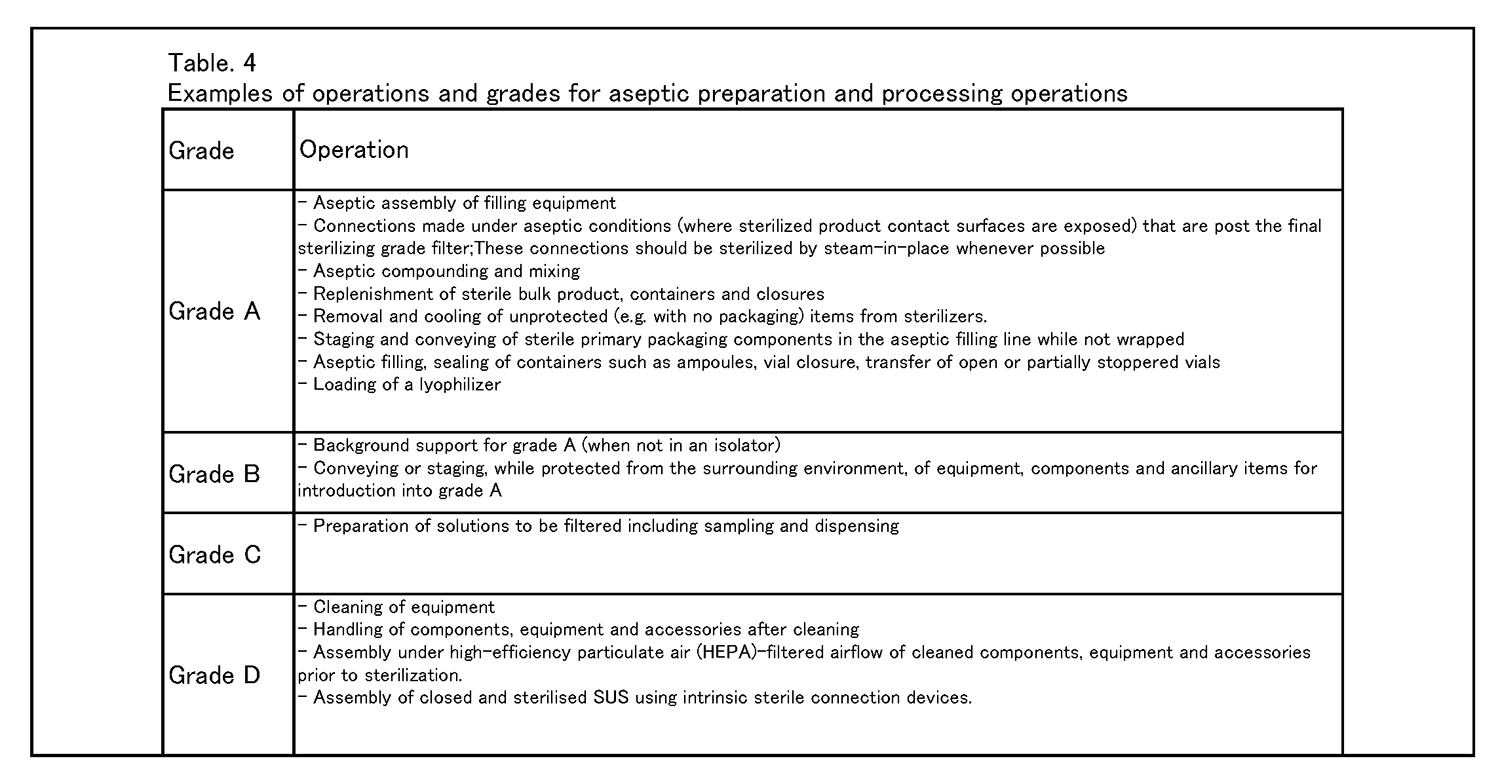 |
8.12 The unwrapping, assembly and preparation of sterilized equipment, components and ancillary items with direct or indirect product contact should be treated as an aseptic process and performed in grade A with a grade B background. The filling line set-up and filling of the sterile product should be treated as an aseptic process and performed in grade A with a grade B background. Where an isolator is used, the background should be in accordance with paragraph 4.20. | 8.13 Preparation and filling of sterile products such as ointments, creams, suspensions and emulsions should be performed in grade A with a grade B background when the product and components are exposed to the environment and the product is not subsequently filtered (via a sterilizing grade filter) or terminally sterilized. Where an isolator or RABS is used, the background should be in accordance with paragraph 4.20. | 8.14 Aseptic connections should be performed in grade A with a grade B background unless subsequently sterilized in place or conducted with intrinsic sterile connection devices that minimize any potential contamination from the immediate environment. Intrinsic sterile connection devices should be designed to mitigate risk of contamination. Where an isolator is used, the background should be in accordance with paragraph 4.20. Aseptic connections should be appropriately assessed and their effectiveness verified (for requirements regarding intrinsic sterile connection devices, refer to paragraphs 8.129 and 8.130). |
9 Environmental & process monitoring Environmental monitoring - total particle 9.9 The appropriate alert limits and action limits should be set for the results of viable and total particle monitoring. The maximum total particle action limits are described in Table.5 and the maximum viable particle action limits are described in Table.6. |
9.12 The monitoring of grade C and D cleanrooms in operation should be performed based on data collected during qualification and routine data to allow effective trend analysis. The requirements of alert limits and action limits will depend on the nature of the operations carried out. Action limits may be more stringent than those listed in Tables.5 and.6 below. Environmental monitoring : total Particle 9.15 The limits for environmental monitoring of airborne particle concentration for each graded area are given in Table 5. |
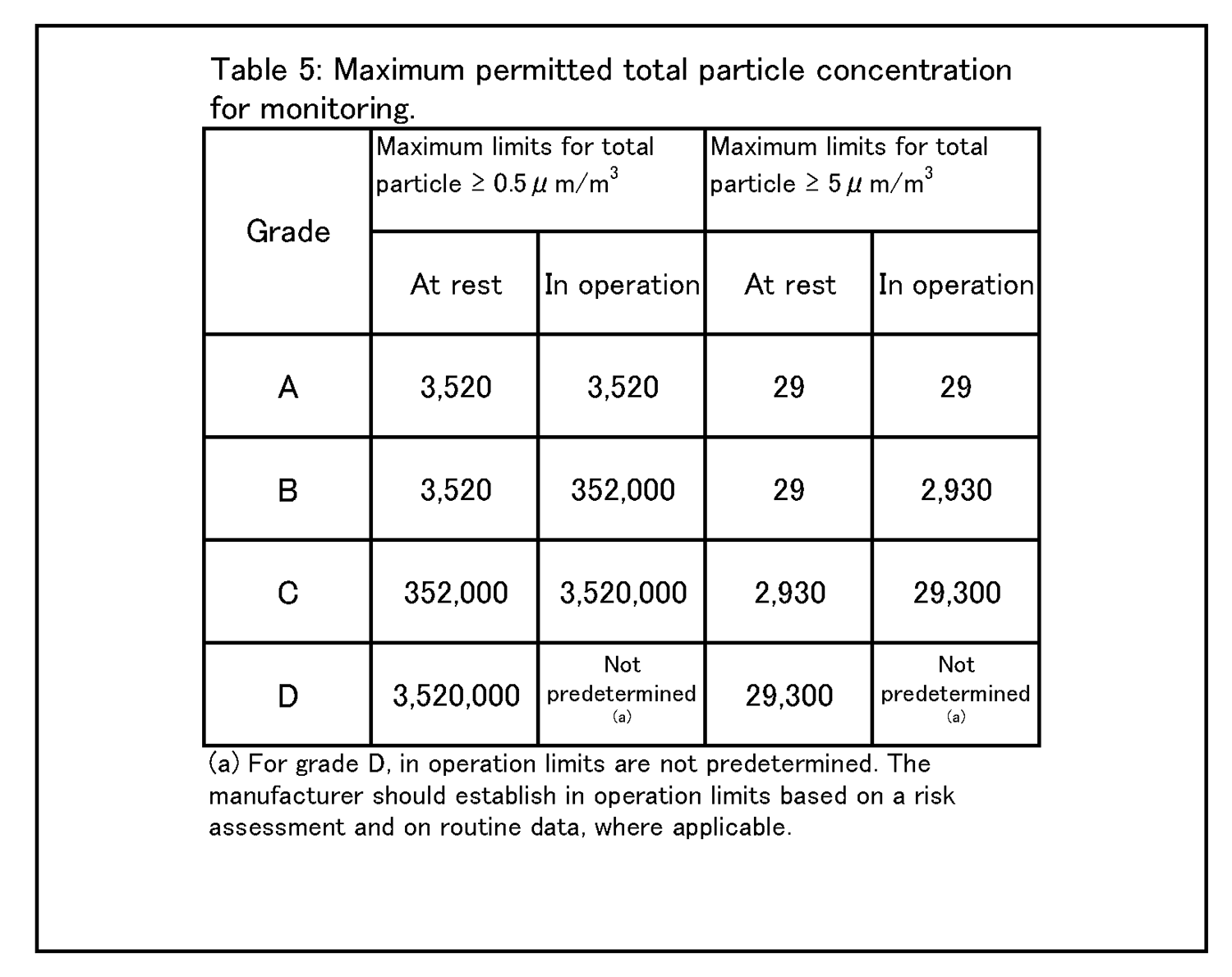 |
Note 1: The particle limits given in the table for the at rest state should be achieved after a short clean-up period defined during qualification (guidance value of less than 20 minutes) in an unmanned state, after the completion of operations (refer to paragraph 4.29). Note 2: The occasional indication of macro particle counts, especially ≧ 5 μm, within grade A may be considered to be false counts due to electronic noise, stray light, coincidence loss, or other factor. However, consecutive or regular counting of low levels may be indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the room air supply filtration system or equipment failure, or may be diagnostic of poor practices during machine set-up and routine operation. |
Glossary isolator. An enclosure capable of being subject to reproducible interior biodecontamination, with an internal work zone meeting grade A conditions that provide uncompromised continuous isolation of its interior from the external environment (for example, surrounding cleanroom air and personnel). There are two major types of isolators: ■ Closed isolator systems exclude external contamination of the isolator’s interior by accomplishing material transfer via aseptic connection to auxiliary equipment rather than use of openings to the surrounding environment. Closed systems remain sealed throughout operations. |
(続き) ■ Open isolator systems are designed to allow for the continuous or semicontinuous ingress or egress of materials during operations through one or more openings. Openings are engineered (for example, using continuous overpressure) to exclude the entry of external contaminant into the isolator |
Glossary restricted access barrier system (RABS). A system that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A) and using a rigid wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, rapid transfer systems or ports, and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened and only under strictly predefined conditions. |
Glossary rapid transfer system or port. A system used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Glossary airlock. An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a less controlled area. |
Glossary pass-through hatch. Synonymous with airlock [refer to airlock definition] but typically smaller in size. |
4. Premises 4.1 Premises design Manufacturers may use different terms when classifying areas, including Grade A, B, C, D, or ISO 7, ISO 8, or Level 1, Level 2 or others (5) (see Table A2.1). When classifying an area, the class selected should be defined and described (see also Section 7). Table A2.1 Examples of area classification (5) (表は省略) |
5. Design of HVAC systems and components The HVAC system should be appropriately designed, taking into consideration the design of the facility, with various rooms or areas for storage of materials and inprocess materials or products, processing, and movement of materials, products and personnel. The required cleanliness classification should be achieved, as well as other parameters, such as air filtration, airflow velocity, air volumes, pressure differentials, temperature, relative humidity, viable and non-viable particle counts and containment. | (続き) Conditions and limits should be specified, based on need. Manufacturers should determine and define limits for these.These should be realistic, appropriate and scientifically justifiable at rest, in operation and as built at the time of design.In determining these, relevant factors and risks should be considered, including but not limited to possible failures of AHUs, seasonal variations, properties and types of materials and products, numbers of personnel and risks of cross-contamination. |
| 概 要 | 対象医薬品:無菌医薬品 以下の要件があります。 ・作業室又は作業管理区域の清浄度の維持管理に関する手順書について。 |
対象医薬品:無菌医薬部外品 以下の要件があります。 ・作業室又は作業管理区域の清浄度の維持管理に関する手順書について。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・作業時、非作業時それぞれの状態における許容微粒子数を規定しています。また0.5μm以上と5.0μm以上の2つの粒子径についての規定。 ・作業時のグレードDについては作業形態により定める(規定しないケースもある)。 *「作業時、非作業時」の定義の記載はありません。 本表、PIC/S GMP ガイドライン アネックス1 4.29に定義の記載があります。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・作業時、非作業時それぞれの状態における許容微粒子数を規定しています。また0.5μm以上と5.0μm以上の2つの粒子径についての規定。 ・作業時のグレードDについては作業形態により定める(規定しないケースもある)。 *「作業時、非作業時」の定義の記載はありません。 本表、PIC/S GMP ガイドライン アネックス1 4.29に定義の記載があります。 |
対象医薬品: 全般 以下の要件があります。 ・各室のレイアウトにおいては製造区域において作業の流れと必要な清浄度を論理的に構築できように考慮すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・清浄度区分のグレードA区画の説明と局所的な気流保護の実証と評価について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードA区域内の介入操作を最小化する設計について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・清浄度区分のグレードB、C、Dの区画説明と例、グレードAおよびアイソレータのバックグラウンドの場合について。 *用語ついての補足:「アイソレータ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・異なるグレード間の出入りに関するエアロックの清浄度設定、グレードBの更衣室の構成について。 *用語ついての補足:「エアロック」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・エアロックの用途と設置と清浄度区分例、更衣室の手洗いの設置について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードAで使用する物/設備がグレードB区域を通過する場合について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・パススルーハッチ自体のフィルター給気による高い清浄度の環境維持と、搬送する物/設備の清浄化、消毒。 *用語ついての補足:「パススルーハッチ」は本表アネックス1の用語解説を参照ください。 「パスボックス」が「パススルーハッチ」に該当します。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・開口部のあるアイソレータの設置環境は最低限グレードC相当であること。 ・閉鎖式のアイソレータの設置環境は最低限グレードD相当であること。 ・RABSの設置環境は最低限グレードB相当であること。 *用語ついての補足:「アイソレータ」は本表アネックス1の用語解説を参照ください。 *用語ついての補足:「RABS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームと空調は適格性評価が必要で、運用時環境モニタリングと区別すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの等級分けに関する適格性評価手法について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの等級ついて、0.5μ m 及び5μm 以上の微粒子の総微粒子量の上限について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの等級および検体採取箇所と数については最低限の基準としてISO14644 パート1によること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの等級における非作業時、作業時の定義について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームのクリーンアップついて、説明と適格性評価の際に決定。 ・クリーンアップ期は20分未満。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微生物、エンドトキシン/発熱性物質及び微粒子のリスクを考慮し、清浄度区分のグレードC、Dの区画で行うべき作業について。 【誤記訂正】厚生労働省通知の和訳では”発別性物質”と記載されていますが、PIC/Sの原文は”pyrogen”であるため、誤記です。表は正しい”発熱性物質”を記載。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・清浄度区分のグレードCの区画で行うべき作業について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・最終滅菌法について、清浄度区分のグレードA、C、D区画で行うべき作業の例。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌操作法について、清浄度区分のグレードA、B、C、D区画で行うべき作業の例。 【誤記訂正】厚生労働省通知の和訳では表4の左列最下段に ""グレードC"" と記載されていますが、PIC/Sの原文はGradeDであるため、誤記です。表は正しいグレードDを記載。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌操作工程の説明と、清浄度区分のグレードA、B区画で行うべき作業の例。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・調製作業及び容器充填作業の製品及び構成物が環境中に露出される場合について、清浄度区分のグレードA、B区画で行うべき作業の例。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌接続について、清浄度区分のグレードA、B区画で行うべき作業の例。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・生菌粒子及び総微粒子量のモニタリングに対する警報基準値及び処置限度値。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・生菌粒子及び総微粒子量のモニタリングに対する警報基準値及び処置限度値が設定について。 ・モニタリングでの総微粒子量の許容上限 ・グレードAのモニタリングに対する説明について。 【誤記訂正】厚生労働省通知の和訳では表5 の最右列、対象粒径5μのグレードB 作業時について""2 9300""と記載されていますが、PIC/Sの原文は""2 930""であるため、誤記です。表は正しい値 2,930を記載。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アイソレータの定義。 ・ 閉鎖式アイソレータシステムの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アイソレータの定義。 ・ 開口式アイソレータシステムの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アクセス制限バリアシステム(RABS)の定義。 *用語ついての補足:「RTP」は本表アネックス1の用語解説の「迅速搬送システム/ポート(RTP)」を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・迅速搬送システム/ポート(RTP)の定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・エアロックの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・パススルーハッチの定義。 |
対象医薬品:ヒト用先端医療医薬品の製造 以下の要件があります。 ・封じ込め目的で陰圧管理をおこなう場合の清浄グレードの陽圧管理ゾーンの設置について。 |
対象医薬品:ヒト用先端医療医薬品の製造 以下の要件があります。 ・原料や製品のリスクを考慮した、製造建屋の微粒子及び微生物環境管理のグレードの設定。 ・閉鎖系でなく微生物不活化処理を経ずに製品が直接室内環境に露出するプロセスにおける無菌操作でのグレードの設定。 |
対象医薬品:ヒト用先端医療医薬品の製造 以下の要件があります。 ・閉鎖システムにおける清浄度区分のグレードの設定。 ・QRM評価の結果に基づき、製品のリスク等を考慮して、等級の設定をすることについて。 |
対象医薬品:ヒト用先端医療医薬品の製造 以下の要件があります。 ・グレードAより低い設定にする例。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) の製造 以下の要件があります。 ・製造建屋の微粒子及び微生物環境管理の度合い。 ・封じ込め目的で陰圧管理区域又は安全キャビネットを使用する場合の周囲の清浄グレード。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・作業時の状態における許容微粒子数を規定しています。また0.5μm以上の粒子径についてのみ規定。 *非作業時(as-built, static conditions)の内容についての記述はありません。 *作業時(dynamic condition):作業者がいて装置が据付られており、製造が行われている状態、と記載されています。 |
対象医薬品: 全般 「別紙(1) PIC/S GMP ガイドライン パート1 第3 章 建物及び設備 製造区域 3.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.24と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.26と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.27と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.28と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.29と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.29と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.3,8.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.6と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.13と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.12と同じ内容となります。 環境モニタリング ー 総微粒子量 9.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、閉鎖式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、開口式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品の製造 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.7と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品の製造 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.10、3.11と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品の製造 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.13と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品の製造 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.13と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第3 章 建物及び設備 製造区域 3.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.24と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.26と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.27と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.28と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.29と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.29と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.3、8.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.6と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.13と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.12、9.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、閉鎖式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、開口式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.24と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.26と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.27と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.28と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.29と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.29と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.3,8.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 最終滅菌法による製品 8.6と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.13と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.12と同じ内容となります。 環境モニタリング ー 総微粒子量 9.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、閉鎖式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、開口式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・清浄度の等級分けとしてGrade A, B, C, D、ISO 7, ISO 8、Level 1, Level 2あるいはその他の用語があり、各等級の定義と記述が必要。 ・等級分けの例として表A2.1にて対象となる区域について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・HVACシステムは、施設において、材料や製品の保管、処理、移動のためのさまざまな部屋やエリアを含めて、微粒子及び微生物等のパラメータを考慮し、適切に設計され、必要な清浄度分類を達成すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・必要とされる清浄度等級分け、管理限度値、それらの設定におけるリスクの考慮について。 |
|||||||||||||||||||||||
| 国 | 日本 | 日本 | 日本 | 日本 | 米国 | ― | ― | 欧州 | 欧州 | ― | ― | ― | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | WHO | |||||||||||||||||||||||||||||||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品 の製造に関する指針 |
最終滅菌法による無菌医薬品 の製造に関する指針 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
PIC/SのGMPガイドラインを活用する際の考え方について 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing ー Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS Annex 3 (Manufacture of radiopharmaceuticals) | EU Guidelines to Good Manufacturing Practice
Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines to Good Manufacturing Practice
Medicinal Products for Human and Veterinary Use Annex 3 Manufacture of Radiopharmaceuticals |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
|||||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2011年 | 2012年 | 2025年 | 2012年 | 2004年 | 2023年 | 2023年 | 2022年 | 2008年 | 2022年 | 2018年 | 2019年 | |||||||||||||||||||||||||||||||||||||||||||
| 記載内容 | 6.構造設備 6.1 構造設備の設計上の要点 29) 原料の秤量作業又は容器の洗浄作業を行う部屋は隣接する 他の部屋への影響を考慮し、シール性や気流方向に注意すること。 |
7.無菌医薬品に係る製品の作業所 7.2 空調システム 7.2.2 空気 各清浄区域の環境を維持するためには、清浄度レベルの高い区域から、隣接する清浄度レベルの低い区域へ流れる適切な気流を確保することが重要である。 2) 無菌操作区域とその他の支援区域との間にはエアロックを設け、室間差圧及び気流の逆転が起きないよう、十分な差圧を設けること。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 28)原料の秤量作業又は容器の洗浄作業を行う部屋は、隣接する他の部屋への影響を考慮し、シール性や気流方向に注意すること。 |
6. 最終滅菌法による医薬品の製造区域 6.2 空調システム 6.2.2 空気 各清浄区域の環境を維持するためには、清浄度レベルの高い区域から、隣接する清浄度レベルの低い区域へ流れる適切な気流を確保することが重要である。 |
4 建物 4.14 クリーンルームは、全ての作業条件下で、より低い等級のバックグラウンド環境に比して陽圧及び/又は気流を保持するフィルタ処理された給気が供給されていること、また、その給気は当該区域を効果的に換気するものであること。(略) 封じ込めで重要区域に空気を流入させることを要する場合には、当該空気の供給源は、同等以上の等級の区域からとすること。 |
4.15 クリーンルーム及び清浄区画内での気流パターンを視覚化して、低い等級から高い等級の区域へ流入することがない旨、また、低い等級の区域(床など) から又はより高い等級の区域へ汚染を運ぶおそれのある作業者又は設備の上を空気が漂うことのない旨を、実証すること。 | (続き) (略) 容器充填され且つ閉塞された製品を小さな搬出口を介してより低い等級の隣接クリーンルームへ搬送する際には、空気がより低い等級のクリーンルームからグレードB区域へ入り込むことがない旨を、気流視覚化検討試験で実証すること。空気が動くことで清浄区域又は重要区域に対する汚染リスクになることが判明している場合には、是正措置(設計改良など) を実施すること。気流パターンの検討試験は、非作業時及び作業時(例: 作業者の介在をシミュレートする) の両方が行われていること。気流パターンのビデオ記録が保存されていること。空気の視覚化検討試験の結果を文書化するとともに、その施設の環境モニタリングプログラムを策定する際に検討すること。 |
バリア技術 4.20 アイソレータ又はRABSのバックグラウンド環境は、汚染の伝播するリスクが最小化されていることを確保するものであること。 i.アイソレータ c. 気流パターンの検討試験を開口式アイソレータの境界面で行って、空気が入ってこないことを実証すること。 ii.RABS 無菌操作用のRABS のバックグラウンド環境は最低限グレードB相当であること、また、気流パターンの検討試験を行って、介入操作(該当する場合にはドアを開くことを含む)の際に空気が入ってこないことを実証すること。 |
クリーンルーム及び清浄空気設備の適格性評価 4..31 クリーンルームの微生物汚染レベルが、そのクリーンルームの適格性評価の一部として決定されていること。検体採取箇所の数は、文書化されたリスク評価、及び部屋の等級分け、気流視覚化検討試験及び当該区域内で行われる工程・作業についての知識から得られた結果に基づくものであること。各グレードの適格性評価の際の微生物汚染の最大限度値を、表2に示す。適格性評価には「非作業時」及び「作業時」状態の両方を含めること。(表2 略) |
7人員 7.18 無菌区域における作業のうち製造工程に重要でないものは、無菌作業が進行しているときには特に、最小限にとどめること。人員の動きはゆっくりと、管理されて規則正しいものとし、過度の激しい活動による微粒子及び微生物の極端な拡散を防止すること。無菌作業を実行している作業者は常に無菌操作技術を厳守して、低品質の空気を重要区画内に流入させるおそれのある空気の流れの変化を防止すること。重要区画近辺での動きを制限し、一方向(ファーストエア)気流の経路の妨げとならないようにすること。気流可視化検討試験の照査を、教育訓練プログラムの一環として、検討すること。 |
8 製造及び特有の技術 無菌操作法による調製・工程作業 8.16 生産に際して発生し得る介入操作(9.34 節を参照) であって許容されていて且つ適格性評価済みのもの(本来的な介入操作及び是正介入操作の両者)についての、承認されたリストがあること。介入操作は、環境、工程及び製品の汚染のリスクが効果的に最小化されていることを確保するよう慎重に設計されていること。介入操作を設計するプロセスには、気流及び重要接触面並びに製品へのインパクトについての検討を含めること。 |
用語解説 アイソレータ - 内部に再現性のあるバイオ除染を行うことが可能な筐体で、グレードA条件に合致する内部作業区画を有し、その内部を毀損することなく外部環境(例:クリーンルームの周囲の空気及び人員)から持続的に隔離状態にするもの。アイソレータには主要な2つの種類がある。 (略) ii. 開口式アイソレータシステムは、1以上の開口部があり、作業の際に連続的又は半連続的に原材料を出し入れできるように設計されている。開口部は、外からの汚染物質がアイソレータに入り込まないようにする造り(例:持続的な過圧を用いる)になっている。 |
用語解説 アクセス制限バリアシステム(RABS) - 所定の空気品質条件(無菌操作にはグレードA)に合致する閉鎖された(だが完全密封ではない)環境を提供するシステムで、堅牢な壁で囲われた筐体と一体化した手袋を用いて、周囲のクリーンルーム環境からその内部を分離するシステム。RABSの内表面は殺芽胞剤で消毒及び除染される。作業者は、手袋、半身スーツ、RTPその他の一体化された搬送ポートを使用して、RABS内部に操作を実行したり原材料を搬入したりする。その設計によっては、ドアが開かれることは稀であり、予め厳密に定められた条件下でのみ開けられる。 |
用語解説 迅速搬送システム/ポート(RTP) - RABS又はアイソレータ内への物品搬送用のシステムで、重要区画へのリスクを最小化するもの。一例として、アルファ/ ベータのポートがある迅速搬送容器が挙げられる。 |
無菌製造 26. 放射性医薬品の生産に関しては、適切な差圧、気流の方向、空気の質を決定するために、リスク評価を適用できる。 |
V. Buildings and Facilities C. Clean Area Separation An essential part of contamination prevention is the adequate separation of areas of operation. To maintain air quality, it is important to achieve a proper airflow from areas of higher cleanliness to adjacent less clean areas. It is vital for rooms of higher air cleanliness to have a substantial positive pressure differential relative to adjacent rooms of lower air cleanliness. For example, a positive pressure differential of at least 10-15 Pascals (Pa) should be maintained between adjacent rooms of differing classification (with doors closed). When doors are open, outward airflow should be sufficient to minimize ingress of contamination, and it is critical that the time a door can remain ajar be strictly controlled . |
4 Premises 4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. (略)Where containment requires air to flow into a critical zone, the source of the air should be from an area of the same or higher grade. |
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is no ingress from lower grade to higher grade areas and that air does not travel from less clean areas (such as the floor) or over operators or equipment that may transfer contamination to the higher-grade areas. | (続き) (略) When filled, closed products are transferred to an adjacet cleanroom of a lower grade via a small egress point, airflow visualization studies should demonstrate that air does not ingress from the lower grade cleanrooms to the grade B area. Where air movement is shown to be a contamination risk to the clean area or critical zone, corrective actions, such as design improvement, should be implemented. Airflow pattern studies should be performed both at rest and in operation (e.g. simulating operator interventions).Video recordings of the airflow patterns should beretained. The outcome of the air visualisation studies should be documented and considered whenestablishing the facility's environmental monitoring programme. |
BARRIER TECHNOLOGIES 4.20 The background environment for isolators or RABS should ensure the risk of transfer of contamination is minimized. i. Isolators: c. Airflow pattern studies should be performed at the interfaces of open isolators to demonstrate the absence of air ingress. ii. RABS: The background environment for RABS used for aseptic processing, should correspond to a minimum of grade B and airflow pattern studies should be performed to demonstrate the absence of air ingress during interventions, including door openings if applicable. |
CLEANROOM AND CLEAN AIR EQUIPMENT QUALIFICATION 4.31 The microbial contamination level of the cleanrooms should be determined as part of the cleanroom qualification. The number of sampling locations should be based on a documented risk assessment and the results obtained from room classification, air visualization studies and knowledge of the process and operations to be performed in the area. The maximum limits for microbial contamination during qualification for each grade are given in Table 2. Qualification should include both “at rest” and “in operation” states. (Table 2 略) |
7 Personnel 7.18 Activities in clean areas that are not critical to the production processes should be kept to a minimum, especially when aseptic operations are in progress. Movement of personnel should be slow, controlled and methodical to avoid excessive shedding of particles and organisms due to over-vigorous activity. Operators performing aseptic operations should adhere to aseptic technique at all times to prevent changes in air currents that may introduce air of lower quality into the critical zone. Movement adjacent to the critical zone should be restricted and the obstruction of the path of the unidirectional (first air) airflow should be avoided. A review of airflow visualisation studies should be considered as part of the training programme. |
8 Production and Specific Technologies Aseptic preparation and processing 8.16 There should be an authorized list of allowed and qualified interventions, both inherent and corrective, that may occur during production (see paragraph 9.34). Interventions should be carefully designed to ensure that the risk of contamination of the environment, process and product is effectively minimized. The process of designing interventions should include the consideration of any impact on air-flows and critical surfaces and products. | Glossary Isolator - An enclosure capable of being subject to reproducible interior bio-decontamination, with an internal work zone meeting grade A conditions that provides uncompromised, continuous isolation of its interior from the external environment (e.g. surrounding cleanroom air and personnel). There are two major types of isolators: (略) ii. Open isolator systems are designed to allow for the continuous or semi-continuous ingress and/or egress of materials during operations through one or more openings. Openings are engineered (e.g. using continuous overpressure) to exclude the entry of external contaminant into the isolator. |
Glossary Restricted Access Barrier System (RABS) - System that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A), and using a rigid-wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, RTPs and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened, and only under strictly pre-defined conditions. |
Glossary Rapid Transfer System/Port (RTP) - A System used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Premises and equipment Sterile production 26. For manufacture of radiopharmaceuticals a risk assessment may be applied to determine the appropriate pressure differences, air flow direction and air quality. |
4 Premises 4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. (略) Where containment requires air to flow into a critical zone, the source of the air should be from an area of the same or higher grade. |
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is no ingress from lower grade to higher grade areas and that air does not travel from less clean areas (such as the floor) or over operators or equipment that may transfer contamination to the higher grade areas. | (続き) (略) When filled, closed products are transferred to an adjacent cleanroom of a lower grade via a small egress point, airflow visualization studies should demonstrate that air does not ingress from the lower grade cleanrooms to the grade B area. Where air movement is shown to be a contamination risk to the clean area or critical zone, corrective actions, such as design improvement, should be implemented. Airflow pattern studies should be performed both at rest and in operation (e.g. simulating operator interventions). |
Barrier Technologies 4.20 The background environment for isolators or RABS should ensure the risk of transfer of contamination is minimized. i. Isolators: c Airflow pattern studies should be performed at the interfaces of open isolators to demonstrate the absence of air ingress. ii. RABS: The background environment for RABS used for aseptic processing should correspond to a minimum of grade B and airflow pattern studies should be performed to demonstrate the absence of air ingress during interventions, including door openings if applicable. |
Cleanroom and clean air equipment qualification
4.31 The microbial contamination level of the cleanrooms should be determined as part of the cleanroom qualification. The number of sampling locations should be based on a documented risk assessment and the results obtained from room classification, air visualization studies and knowledge of the process and operations to be performed in the area. The maximum limits for microbial contamination during qualification for each grade are given in Table 2. Qualification should include both “at rest” and “in operation” states. (Table 2 略) |
7 Personnel 7.18 Activities in clean areas that are not critical to the production processes should be kept to a minimum, especially when aseptic operations are in progress. Movement of personnel should be slow, controlled and methodical to avoid excessive shedding of particles and organisms due to over-vigorous activity. Operators performing aseptic operations should adhere to aseptic technique at all times to prevent changes in air currents that may introduce air of lower quality into the critical zone. Movement adjacent to the critical zone should be restricted and the obstruction of the path of the unidirectional (first air) airflow should be avoided. A review of airflow visualisation studies should be considered as part of the training programme. |
8 Production and Specific Technologies
Aseptic preparation and processing 8.16 There should be an authorized list of allowed and qualified interventions, both inherent and corrective, that may occur during production (see paragraph 9.34). Interventions should be carefully designed to ensure that the risk of contamination of the environment, process and product is effectively minimized. The process of designing interventions should include the consideration of any impact on air-flows and critical surfaces and products. |
Glossary Isolator - An enclosure capable of being subject to reproducible interior bio-decontamination, with an internal work zone meeting grade A conditions that provides uncompromised, continuous isolation of its interior from the external environment (e.g. surrounding cleanroom air and personnel). There are two major types of isolators: (略) ii. Open isolator systems are designed to allow for the continuous or semi-continuous ingress and/or egress of materials during operations through one or more openings. Openings are engineered (e.g. using continuous overpressure) to exclude the entry of external contaminant into the isolator. |
Glossary Restricted Access Barrier System (RABS) - System that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A), and using a rigid-wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, RTPs and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened, and only under strictly pre-defined conditions. |
Glossary Rapid Transfer System/Port (RTP) - A System used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Premises and equipment Sterile production 26. For manufacture of radiopharmaceuticals a risk assessment may be applied to determine the appropriate pressure differences, air flow direction and air quality. |
4. Premises 4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. (略) Where containment requires air to flow into a critical zone, the source of the air should be an area of the same or higher grade. |
4.15 Airflow visualization studies should demonstrate airflow patterns within cleanrooms and zones proving that there is no ingress from lower-grade to higher-grade areas and that air does not flow from less clean areas (such as the floor) or over operators or equipment, thus transferring contaminants to the higher-grade areas. | (続き)(略) When filled and closed products are transferred to an adjacent cleanroom of a lower grade via a small exit point, airflow visualization studies should demonstrate that there is no ingress from the lower-grade cleanroom to the grade B area. Where air movement is shown to be a contamination risk to the clean area or critical zone, corrective action, such as design improvement, should be implemented. Airflow pattern studies should be performed both at rest and in operation (for example, simulating operator interventions). |
Barrier technologies 4.20 The background environment for isolators and RABS should ensure that the risk of transfer of contamination is minimized. i. Isolators: c.Airflow pattern studies should be performed at the interfaces of open isolators to demonstrate the absence of air ingress. ii. RABS: a. The background environment for RABS used for aseptic processing should correspond to a minimum of grade B, and airflow pattern studies should be performed to demonstrate the absence of air ingress during interventions, including door openings if applicable. |
4.31 The microbial contamination level of the cleanrooms should be determined as part of the cleanroom qualification. The number of sampling locations should be based on a documented risk assessment and the results obtained from room classification, air visualization studies, and knowledge of the process and operations to be performed in the area. The maximum limits for microbial contamination during qualification for
each grade are given in Table.2. Qualification should include both at rest and operational states. (Table 2 略) |
7. Personnel 7.18 Activities in clean areas that are not critical to the production processes should be kept to a minimum, especially when aseptic operations are in progress. The movement of personnel should be slow, controlled and methodical to avoid excessive shedding of particles and organisms due to overvigorous activity. Operators performing aseptic operations should adhere to aseptic technique at all times to prevent changes in air currents that may introduce air of lower quality into the critical zone. Movement adjacent to the critical zone should be restricted and obstruction of the path of the unidirectional (first air) airflow should be avoided. A review of airflow visualization studies should be considered as part of the training programme. |
8. Production and specific technologies
Aseptic preparation and processing 8.16 There should be an authorized list of allowed and qualified interventions, both inherent and corrective, that may occur during production (refer to paragraph 9.34). Interventions should be carefully designed to ensure that the risk of contamination of the environment, process and product is effectively minimized. The process of designing interventions should include the consideration of any impact on airflows and critical surfaces and products. |
Glossary isolator. An enclosure capable of being subject to reproducible interior biodecontamination, with an internal work zone meeting grade A conditions that provide uncompromised continuous isolation of its interior from the external environment (for example, surrounding cleanroom air and personnel). There are two major types of isolators: (略) ■ Open isolator systems are designed to allow for the continuous or semicontinuous ingress or egress of materials during operations through one or more openings. Openings are engineered (for example, using continuous overpressure) to exclude the entry of external contaminant into the isolator. |
Glossary restricted access barrier system (RABS). A system that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A) and using a rigid wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, rapid transfer systems or ports, and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened and only under strictly predefined conditions. |
Glossary rapid transfer system or port. A system used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
4. Premises 4.10 Detailed schematic diagrams should be maintained, indicating, for example, air supply and air return, room pressure differentials and airflow directions. |
7. Air filtration, airflow direction and pressure differentials 7.1 Where different products are manufactured at the same time, i.e. in different areas or rooms in a multiproduct manufacturing site, measures should be taken to ensure that dust cannot move from one room to another. Facility design and layout, appropriate levels of filtration, airflow direction and pressure differentials can assist in preventing cross-contamination. 7.3 Airflow directions should be appropriate, taking operator and equipment locations into consideration. |
7.4 The pressure differential between areas in a facility should be individually assessed according to the products handled and level of protection required. The pressure differential and the direction of airflow should be appropriate to the product and processing method used, and should also provide protection for the operator and the environment. 7.5 The pressure differential should be designed so that the direction of airflow is from the clean area, resulting in dust containment, for example, from the corridor to the cubicle. |
4. Premises 4.2 Weighing/dispensing and sampling areas (略) Washing areas should be designed and used in such a manner that equipment and components will not be re-contaminated after cleaning. The system supplying and extracting air from the area(s) should be suitably designed to ensure that this objective is achieved. Principles that may be considered include (but are not limited to) filtration of air, pressure differentials between areas, air changes per hour and airflow directions (for an example, see Fig. A2.7). |
5. Design of HVAC systems and components 5.1 Containment Manufacturers should ensure that appropriate measures are taken to contain product dust in a manufacturing area, thus preventing or minimizing the risk of contamination of other areas and possible cross-contamination. In some cases, it may be advisable to have airlocks or pass-through hatches between rooms or areas. In addition, sufficient dilution, pressure differentials (recommended minimum values of 5 Pa) and airflow directions can further support containment in an area. |
7. Air filtration, airflow direction and pressure differentials (略) Airflow directions should be specified and proven to promote containment and not be adversely affected or obstructed by equipment, utilities, containers or personnel. The location of supply and return or exhaust air grilles should facilitate appropriate airflow directions in an area. |
9. Dust, vapour and fume control (略)As vapours can be problematic, extraction may be supported by directional airflow to assist in the removal. (略) |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・秤量作業や洗浄作業による室外への汚染防止手段として気流方向に注意が必要。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・汚染防止手段としての気流の方向の設定。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・秤量作業や洗浄作業による室外への汚染防止手段として気流方向に注意が必要。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・汚染防止手段としての気流の方向の設定。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームとしてバックグラウンド環境に向かう気流の維持や、流入する気流の清浄度等級ついて。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・気流可視化における実証について、 (清浄度区域間の低い等級からの気流の流入防止、汚染を含むおそれのある気流の流入防止) |
対象医薬品:無菌医薬品 以下の要件があります。 ・気流可視化における実証について。 (グレードB区域と隣接する低い等級間の充填閉塞済製品の搬送排出口) ・気流パターンは、非作業時及び作業時の両方が必要で、気流の映像を記録すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・開口式アイソレータ及びRABSにおけるバックグラウンド環境の気流可視化における実証について。 *用語ついての補足:「開口式アイソレータ」は本表アネックス1の用語解説を参照ください。 *用語ついての補足:「RABS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微生物関係の検体採取箇所の数は、気流視覚化検討試験結果に基づくこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌作業の作業者は、低品質の空気を重要区画内に流入させるおそれのある空気の流れの変化を防止すること。 ・教育訓練プログラムとして、気流可視化検討試験結果の確認を含めること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・生産中に作業者が介在する介入操作においては製品汚染のリスクを避けるために気流等について、設計時に考慮すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アイソレータの定義。 ・ 開口式アイソレータシステムの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アクセス制限バリアシステム(RABS)の定義。 *用語ついての補足:「RTP」は本表アネックス1の用語解説の「迅速搬送システム/ポート(RTP)」を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・迅速搬送システム/ポート(RTP)の定義。 |
対象医薬品:放射性医薬品のうち無菌製品 以下の要件があります。 ・気流方向をリスク評価により定められる。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・清浄度設定に対する気流について、またドア開放時の気流について。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14,と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7人員 7.18と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、開口式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:放射性医薬品のうち無菌製品 別紙(2) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 無菌製造 26..と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14,と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7人員 7.18と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、開口式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:放射性医薬品のうち無菌製品 別紙(2) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 無菌製造 26.と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7人員 7.18と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、開口式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・気流方向を含む詳細な設計図が管理され、示めすこと。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・異なる製品を同時製造する場合の製造室間の気流方向。 ・気流方向の設定における作業者や装置の位置との関係について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・塵埃の拡散を抑制する気流方向。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・洗浄室エリアの空調について清浄化済みの装置や部品が再汚染しないよう気流方向を設定すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・製品自体の粒子を封じ込め、他の区域の汚染および交叉汚染を防ぎ、リスクを最小とするために気流方向等を設定すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・気流方向における設定と検証について。 ・作業室の給気および吸込口の配置と気流方向。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・有害な蒸発由来物質と集塵における気流方向。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | 米国 | ― | ― | ― | 欧州 | 欧州 | ― | ― | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CDER | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | |||||
| 分 類 | 省令 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長 | Code of Federal Regulations | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 | 薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2A ヒト用先端医療医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2B ヒト用生物学的医薬品( 原薬及び製剤) の製造 |
「PIC/S のGMP ガイドラインを活用する際の考え方について」の一部改正について 別紙(13) PIC/S GMP ガイドライン アネックス14 ヒト血液及びヒト血漿由来製品の製造 |
原薬GMPのガイドラインについて | TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing ― Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS
ANNEX 2A MANUFACTURE OF ADVANCED THERAPY MEDICINAL PRODUCTS FOR HUMAN USE |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS
ANNEX 2B MANUFACTURE OF BIOLOGICAL MEDICINAL SUBSTANCES AND PRODUCTS FOR HUMAN USE |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 14 Manufacture of products derived from human blood or human plasma |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 54th report Annex 2 International Atomic Energy Agency and World Health Organization guideline on good manufacturing practices for radiopharmaceutical products WHO Technical Report Series, No. 1025, 2020 |
|||||
| 発行年 | 2021年 | 2021年 | 2011年 | 2020年 | 2022年 | 2022年 | 2015年 | 2001年 | 2023年 | 2004年 | 2021年 | 2023年 | 2023年 | 2015年 | 2011年 | 2019年 | 2020年 | |||||
| 記載内容 | (品質管理) 第十一条 製造業者等は、品質部門に、手順書等に基づき、次に掲げる品質保証及び試験検査に係る業務を計画的かつ適切に行わせなければならない。 五 最終製品(ロットを構成するものに限る。)について、ロットごとに所定の試験検査に必要な量の二倍以上の量を参考品として、製造された日から当該製品の有効期間に一年(放射性医薬品の最終製品にあっては、六月又は品質リスクマネジメントに基づく適切な日数)を加算した期間適切な保管条件の下で保管すること。また、その保存品を当該参考品と同期間保管すること。 |
六 医薬品に係る製品の製造に使用した原料等のうち当該製品の品質に影響を及ぼすものについて、原料にあってはロットごとに所定の試験検査に必要な量の二倍以上の量を、資材にあっては管理単位ごとに所定の試験検査に必要な量を、それぞれ参考品として、当該製品の出荷を判定した日から二年間(放射性医薬品に係る製品の原料にあっては、当該原料の安定性に基づく適切な期間)適切な保管条件の下で保管すること。 | (特定生物由来医薬品等の製造業者等の製造所の構造設備) 第八条 一 特定生物由来医薬品等に係る製品の製造所(包装、表示又は保管のみを行う製造所を除く。)は、次に定めるところに適合するものであること。 ヲ 貯蔵設備は、恒温装置、自記温度計その他必要な計器を備えたものであること。 |
10. 無菌中間製品の保管及び輸送の管理 10.4 保管及び輸送の条件 温度,環境圧力,振動等無菌性を維持したまま保管及び輸送を行う上でリスクとなる事項に係る条件をあらかじめ設定するとともに,設定した範囲内で保管及び輸送を実施すること. |
建物 3.3. 照明、温度、湿度及び換気が適切であり、製造及び貯蔵中の医薬品並びに装置の正確な作動のどちらにも、それらが( 直接的又は間接的に) 悪影響を及ぼさないこと。 保管区域 3.19. 貯蔵区域は、良好な保存条件を確保するよう設計又は構成すること。特に、当該区域は、清潔で湿気のない状態とし、許容される温度限度値内に保守管理すること。特別な保存条件( 例えば温度、湿度) が求められる場合は、それら条件を規定し、チェックし、モニターすること。 |
パートA : 一般的ガイダンス P I C / S のG M P ガイドライン パートⅠ に補足の条項 第5 章 製造 出発原料として使用されるヒト血液、組織及び細胞 5.36 貯蔵容器は密封し、明確に表示し、適切な温度に保つこと。出納記録を保管しなければならない。貯蔵温度( 及び液体窒素を使用する場合には、その液量)を、継続的にモニターすること。所定の限度値からの逸脱並びに講じられた是正措置及び予防措置を記録すること。 |
パートA . 一般的ガイダンス シードロット及びセルバンクシステム 43. シードロット及びセルバンクは、汚染又は変質のリスクを最小化するようにする方法で貯蔵し( 例: 密封容器内で液体窒素の気相中に貯蔵) 、使用すること。(略) 45. 貯蔵容器は密封し、明確に表示し、適切な温度に保つこと。出納記録を保管しなければならない。貯蔵温度を継続的に記録し、液体窒素を使用する場合には、その液量をモニターすること。所定の限度値からの逸脱並びに講じられた是正措置及び予防措置を記録すること。 |
作業原則 64. 超低温で貯蔵するものについては、当該貯蔵温度でのラベルの適性を検証すること。 |
6.製造 6.6 分画プラントへの輸送チェーンにおけるいかなる段階での血液及び血漿の保管及び輸送を規定し記録すること。 規定された温度からのいかなる逸脱も分画プラントに知らせること。適格性確認された装置及びバリデートされた手順書を用いること。 |
10 保管及び出荷 10.1 保管作業 10.10 全ての原材料等を適切な条件(例えば、必要な場合には管理された温度及び湿度)で保管できる設備を備えること。当該条件が原材料等の特性の維持のために重要な場合には、当該条件の記録を保存すること。 |
Subpart C--Buildings and Facilities Sec. 211.46 Ventilation, air filtration, air heating and cooling. (b) Equipment for adequate control over air pressure, micro-organisms, dust, humidity, and temperature shall be provided when appropriate for the manufacture, processing, packing, or holding of a drug product. |
Subpart H--Holding and Distribution
Sec. 211.142 Warehousing procedures. Written procedures describing the warehousing of drug products shall be established and followed. They shall include: (b) Storage of drug products under appropriate conditions of temperature, humidity, and light so that the identity, strength, quality, and purity of the drug products are not affected. |
Subpart K--Returned and Salvaged Drug Products
Sec. 211.208 Drug product salvaging. Drug products that have been subjected to improper storage conditions including extremes in temperature, humidity, smoke, fumes, pressure, age, or radiation due to natural disasters, fires, accidents, or equipment failures shall not be salvaged and returned to the marketplace. |
IV. BUILDINGS AND FACILITIES 21 CFR 211.46(b) states that “Equipment for adequate control over air pressure, micro-organisms, dust, humidity, and temperature shall be provided when appropriate for the manufacture, processing, packing, or holding of a drug product.” |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES General 3.3.Lighting, temperature, humidity and ventilation should be appropriate and such that they do not adversely affect, directly or indirectly, either the medicinal products during their manufacture and storage, or the accurate functioning of equipment. Storage Areas 3.19. Storage areas should be designed or adapted to ensure good storage conditions. In particular, they should be clean and dry and maintained within acceptable temperature limits. Where special storage conditions are required (e.g. temperature, humidity) these should be provided, checked and monitored. |
Part A. GENERAL GUIDANCE CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Seed Lot and Cell Bank System 5.36 Storage containers should be sealed, clearly labelled and kept at an appropriate temperature. A stock inventory must be kept. The storage temperature and, where used, the liquid nitrogen levels should be continuously monitored. Deviation from set limits and corrective and preventive action taken should be recorded. |
Part A. GENERAL GUIDANCE SEED LOT AND CELL BANK SYSTEM 43. Seed lots and cell banks should be stored and used in such a way as to minimize the risks of contamination (e.g. stored in the vapour phase of liquid nitrogen in sealed containers) or alteration. 45. Storage containers should be sealed, clearly labelled and kept at an appropriate temperature. A stock inventory must be kept. The storage temperature should be recorded continuously and, where used, the liquid nitrogen level monitored. Deviation from set limits and corrective and preventive action taken should be recorded. |
OPERATING PRINCIPLES 64. The compatibility of labels with ultra-low storage temperatures, where such temperatures are used, should be verified. |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES General 3.3.Lighting, temperature, humidity and ventilation should be appropriate and such that they do not adversely affect, directly or indirectly, either the medicinal products during their manufacture and storage, or the accurate functioning of equipment. Storage Areas 3.19. Storage areas should be designed or adapted to ensure good storage conditions. In particular, they should be clean and dry and maintained within acceptable temperature limits. Where special storage conditions are required (e.g. temperature, humidity) these should be provided, checked and monitored. |
6.Manufacturing Starting material 6.6 The storage and transport of blood or plasma at any stage in the transport chain to the fractionation plant should be defined and recorded. Any deviation from the defined temperature should be notified to the fractionation plant. Qualified equipment and validated procedures should be used. |
8. Temperature and relative humidity Manufacturers should set appropriate upper and lower limits for temperature and relative humidity for different areas. The required storage conditions specified for the materials and products should be considered when the limits are defined. |
9. Premises
9.2 Lighting, heating, ventilation and air-cond itioning (HVAC) systems should be designed to maintain an appropriate temperature and relative humidity where required, in order to ensure the appropriate equipment performance, material storage, safety and comfort of personnel. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・ロットを構成する最終製品の”参考品”保管条件について。 *「適切な保管条件」の具体的な温度の数値表記はありません。 |
対象医薬品:全般 以下の要件があります。 ・製品の製造に使用した原料等のうち当該製品の品質に影響を及ぼすものについての適切な保管条件。 *「適切な保管条件」の具体的な温度の数値表記はありません。 |
対象医薬品:特定生物由来医薬品 以下の要件があります。 ・製品の製造所の貯蔵設備には恒温装置、自記温度計その他必要な計器を備えること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・保管および輸送の条件として温度その他の設定と、その維持ができること。 *「温度設定」の具体的な温度の数値表記はありません。 |
対象医薬品:全般 以下の要件があります。 ・保管中の温度が適切であり、製品等に悪影響を及ぼさないこと、および許容される温度限度値内に維持すること、モニタリングを行うこと。 *「保存条件」の具体的な温度の数値表記はありません。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・貯蔵容器の温度を継続的にモニタリングすること、および逸脱時の対応について。 *「貯蔵容器の温度」の具体的な温度の数値表記はありません。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 以下の要件があります。 ・シードロット及びセルバンクについて、貯蔵容器の温度を継続的にモニタリングすること、および逸脱時の対応について。 *「貯蔵容器の温度」の具体的な温度の数値表記はありません。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 以下の要件があります。 ・超低温の保管における容器ラベルについて。 *「超低温」の具体的な温度の数値表記はありません。 |
対象医薬品:ヒト血液及びヒト血漿由来製品 以下の要件があります。 ・分画プラントへの輸送チェーンにおける温度逸脱について。 |
対象医薬品:原薬 以下の要件があります。 ・全ての原材料等における保管の条件の例。温度及び湿度管理。 *「適切な保管条件」の具体的な温度の数値表記はありません。 |
対象医薬品:全般 以下の要件があります。 ・製造や保管時において、温度等を適切に管理する装置を設置。 |
対象医薬品:全般 以下の要件があります。 ・温度等の適切な条件下で保管するなどを記載した倉庫保管手順書。 *「適切な保管条件」の具体的な温度の数値表記はありません。 |
対象医薬品:全般 以下の要件があります。 ・異常事態による極端な温度、不適切な保管条件にさらされた医薬品が使用可能とされ市場に出ることがないこと。 |
対象医薬品:無菌操作法による無菌医薬品 21 CFR 211.46(b)と同内容になります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 建物 3.3. 保管区域 3.19と同じ内容になります。 |
対象医薬品:対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス PIC/S GMP ガイドライン アネックスパートⅠ に補足の条項 第3 章 建物及び設備 建物 シードロット及びセルバンクシステム 5.36と同じ内容になります。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤) の製造 パートA 一般的ガイダンス シードロット及びセルバンクシステム 43、45と同じ内容になります。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤) の製造 パートA 一般的ガイダンス 作業原則 64と同じ内容になります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 建物 3.3と同じ内容となります。 |
対象医薬品:ヒト血液及びヒト血漿由来製品 別紙(13) PIC/S GMP ガイドライン アネックス14 ヒト血液及びヒト血漿由来製品の製造 6.製造 6.6と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・原材料や製品によって定められている保管条件となるように、温度や湿度の上下限値を定めること。 *「保管条件」の具体的な温度の数値表記はありません。 |
対象医薬品:放射線医薬品 以下の要件があります。 ・空調システム等は製品の保管等において適切な温度等を維持するような設計とすること。 *「適切な温度」の具体的な温度の数値表記はありません。 |
| 国 | 日本 | 日本 | 日本 | 米国 | 米国 | ― | 欧州 | ― | ― | ― | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CDER | PIC/S | EUROPEAN COMMISSION | WHO | WHO | WHO | ||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Code of Federal Regulations | Guidance | GMP | GMP | GMP | GMP | GMP | ||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice |
ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS TO GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Usebr>br> Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
||||
| 発行年 | 2011年 | 2012年 | 2025年 | 2023年 | 2004年 | 2023年 | 2022年 | 2022年 | 2018年 | 2019年 | ||||
| 記載内容 | 6.構造設備 6.1 構造設備の設計上の要点 19) 清浄区域にはそこで行われる作業に対して適切な清浄度レベルを維持するためHEPAフィルター等の適切なフィルターによりろ過した空気を供給し,適切な室間差圧を設けること.また,室間差圧が維持されていることを監視できるようにすること. |
5. 建物及び施設 5.1 構造設備の設計上の要点 18) 清浄区域にはそこで行われる作業に対して適切な清浄度レベルを維持するためHEPAフィルター等の適切なフィルターによりろ過した空気を供給し,適切な室間差圧を設けること.また室間差圧が維持されていることを監視できるようにすること. |
8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10 種々の環境清浄度等級において行われる作業の具体例を、表4に示す。 表4:無菌操作法による調製・工程作業のための清浄度等級及び作業の具体例 グレードD (略) - 滅菌前の清浄化済み構成物、設備及びアクセサリー類の、HEPAフィルタ処理済み気流の下での組立て作業 |
SUBPART C BUILDINGS AND FACILITIES § 211.42 Design and construction features (10) Aseptic processing, which includes as appropriate: (iii) An air supply filtered through high-efficiency particulate air filters under positive pressure, regardless of whether the flow is laminar or nonlaminar; |
IV. BUILDINGS AND FACILITIES HEPA-filtered air should be supplied in critical areas at a velocity sufficient to sweep particles away from the filling/closing area and maintain unidirectional airflow during operations. |
8 Production and Specific Technologies Aseptic preparation and processing 8.10 Examples of operations to be carried out in the various environmental grades are given in Table 4. Table 4: Examples of operations and grades for aseptic preparation and processing operations Grade D (略) ‐Assembly under HEPA filtered airflow of cleaned components, equipment and accessories prior to sterilisation. |
8. Production and specific technologies Aseptic preparation and processing 8.10 Examples of operations to be carried out in the various environmental grades are given in Table 4. Table 4: Examples of operations and grades for aseptic preparation and processing operations Grade D (略) - Assembly under HEPA filtered airflow of cleaned components, equipment and accessories prior to sterilisation. |
8. Production and specific technologies Aseptic preparation and processing 8.10 Examples of operations to be carried out in the various environmental grades are given in Table 4.Table. 4 Examples of operations and grades for aseptic preparation and processing operations Grade D (略) - Assembly under high-efficiency particulate air (HEPA)-filtered airflow of cleaned components, equipment and accessories prior to sterilization. |
6. Full fresh air systems and recirculation systems 6.2 HEPA filters may be installed (in the supply air stream or return air stream) to remove contaminants and thus prevent cross-contamination. The HEPA filters in such an application should have an EN 1822 classification of at least H13 or equivalent. 6.3 HEPA filters may not be required to control cross-contamination where evidence that cross-contamination would not be possible has been obtained by other robust technical means, or where the air-handling system is serving a single product facility. |
13. Maintenance 13.6 HEPA filters should be changed by a competent person and this should be followed by installed filter leakage testing. |
6. Full fresh air systems and recirculation systems (略) Manufacturers should ensure that when high-efficiency particulate air (HEPA) filters are used, these are appropriately installed, not damaged and thus suitable for the intended use (see tests described in Section 12). |
7. Air filtration, airflow direction and pressure differentials (略) Filters selected for air filtration should be determined and specified. When a manufacturer chooses to install HEPA filters to achieve the desired degree of filtration of air, these filters may be placed in the AHU, or may be installed terminally near the supply grille. Filters have an impact on the cleanroom class or level of protection. The different levels of protection and recommended filter grades are presented in Table A2.2. |
Table A2.2 Levels of protection and recommended filtration (略) Level 3 Production facility operating on recirculated plus ambient air, where potential for cross-contamination exists: primary plus secondary plus tertiary filters (e.g. EN 779 G4 plus F8 plus EN 1822 H13 filters; for full fresh air system, without recirculation, G4 and F8 or F9 filters are acceptable) |
(続き) Fig. A2.9 is a schematic diagram of an example of an air-handling system serving rooms with horizontal directional flow, vertical directional flow and turbulent flow, for rooms 3, 4 and 5, respectively. In these rooms, the HEPA filters are indicated to have been placed terminally mounted in the rooms and not in the AHU. Whenever HEPA filters are terminally mounted, it can assist with preventing cross-contamination from room to room in the event of a fan failure. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・清浄区域における清浄度レベルを維持するためにHEPAフィルター等の適切なフィルターを設ける。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・清浄区域における清浄度レベルを維持するためにHEPAフィルター等の適切なフィルターを設ける。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードDの加工例として、滅菌工程に先立って行う部品組み立て加工を、HEPAフィルタ気流下で行う。 【誤記訂正】厚生労働省通知の和訳では表4の最下段を”グレードC”と記載していますが、PIC/Sの原文はGrade Dであるため、誤記です。記載内容欄では正しい”グレードD”として記載しました。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 item no.003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌製造におけるHEPAフィルタを通しての給気。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・重要区域(滅菌済み製品や資材などのの滅菌状態を保持しなければならない区域)にはHEPAフィルタによる給気を行うこと。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 無菌操作法による調製・工程作業 8.10と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・ダクト系における交叉汚染防止のためのHEPAフィルタ設置位置について。 ・HEPAフィルタはEN1822規格のH13以上の性能とする。 ・他の手法で交叉汚染を防止できる場合、あるいは空調系統が単一製品向けの場合、HEPAフィルタは必須ではない。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・HEPAフィルタの交換作業は適格な者により行われなければならず、また据付におけるリーク試験も合わせて行わなければならない。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・HEPAフィルタは、損傷しないように、かつ意図した目標に合致するように適切に設置すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・ろ過性能上で必要なフィルタを選定し、仕様規定しなければならない。HEPAフィルタを必要とする場合には、空調機内もしくは給気吹出口に設置される。 ・フィルタはクリーンルームの清浄度管理レベルに影響を与える。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・表A2.2に管理レベルごとの推奨フィルタについて示されており、Level3*1において再循環をする空調システムの3次フィルタとしてEN 1822 H13 filters*2が挙げられている。 *1:「空調 003:クリーンルーム清浄度の設定」 WHO GMP 53rd report Annex2 表2.1参照 *2:WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 6.3参照 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・Fig.A2.9に示す循環式の空調システムにおいては、HEPAフィルタを末端(吹出口)に設置することで、送風機故障時の室間の交差汚染防止となる。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | ― | ― | 欧州 | 欧州 | ― | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | GMP | GMP | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2A ヒト用先端医療医薬品の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Part 1 |
EU Guidelines for Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2011年 | 2012年 | 2020年 | 2025年 | 2022年 | 2004年 | 2021年 | 2023年 | 2014年 | 2022年 | 2022年 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 記載内容 | 7.無菌医薬品に係る製品の作業所 7.1.1 重要区域(グレードA) 4) 製品の無菌性を確保する上で特に重要な箇所については,浮遊微粒子数及び微生物数について適切な方法及び頻度によりモニタリングを行うこと. 浮遊微粒子数は,滅菌した接液パーツの組立て作業など重要な準備作業を含め無菌操作を行っている時間を通して連続的に測定することが望ましい.また,作業域にできるだけ近い位置で(30cm以内が望ましい),測定を行うこと. 7.1.2 直接支援区域(グレードB) 3) 直接支援区域においては浮遊微粒子数及び微生物について定期的にモニタリングを行うこと. その頻度と方法については,重要区域内の製品に対する汚染リスクを評価し,適切に定めること |
11. 環境モニタリング 11.1 一般要求事項 3) モニタリングの対象物 モニタリングの対象物は微生物及び浮遊微粒子とする (ア) 微粒子は粒径0.5μm以上の浮遊微粒子とする.環境モニタリングをより適切に行うために,必要に応じて,適宜,他の粒子径(例:5μm以上)の計測を行う. 11.3 環境モニタリング判定基準例 6) グレードA及びグレードBにおける微粒子管理は,機器の組み立てから重要作業終了までは連続モニタリングを推奨する. |
8. 環境モニタリング 8.1 一般要求事項 3) モニタリングの対象物 モニタリングの対象物は,微生物及び浮遊微粒子とする ① 微粒子は,粒径0.5μm以上の浮遊微粒子とする.環境モニタリングをより適切に行うために,必要に応じて,適宜,他の粒子径(例:5.0μm以上)の計測を行う. 7) モニタリングの方法:試料採取方法及び検出方法 製造区域毎のモニタリングポイントは,作業室の大きさ,作業内容,原材料や製品の工程フローなどを考慮して,適切な分布と採取箇所数を定めること.製品汚染評価に重要と考えられるポイントは適宜追加すること. ① 浮遊微粒子の測定装置及び浮遊微生物の採取装置は,バリデートされた校正済装置を使用すること.微粒子の評価は,適切なサンプリング量を1m3当たりに換算して評価すること |
8.3 環境モニタリング判定基準例 5) グレードA及びグレードBにおける微粒子管理は,機器の組み立てから重要作業終了までは連続モニタリングを推奨する. 6) 製造が行われていない時間帯の微粒子モニタリングは,空調の不具合発見など,環境維持継続性の観点から適宜実施すること. 7) 微粒子の計測については,サンプル量及び吸引能力により評価判定が異なるので,適切な評価ができるような機器及び評価方法によること. |
第5 章 製造 製造における交叉汚染の防止 5.21 品質リスクマネジメントのプロセスの結果に基づいて、交叉汚染に係るリスクを管理するため求められる技術的措置及び組織的措置の範囲を確定させること。 技術的措置及び組織的措置には以下が含まれ得るが、これらに限らない。 組織的措置 i v . 汚染リスクに応じて、空気浮遊物汚染又は機械的移動による汚染に対する管理措置の有効性を実証するために、製品非接触面の清浄化の検証、並びに製造区域及び/ 又は隣接区域内における空気のモニタリングを行う。 |
2 原則 2.1 無菌製品の製造は、微生物、微粒子及びエンドトキシン/発熱性物質の汚染のリスクを最小化するために、特別な要求事項の対象となっている。以下の主要分野が検討されていること。 i . 施設、設備及び工程が、適切に設計され、適格性評価及び/又はバリデートされていて、且つ該当する場合には、GMPガイドラインの関連する項に従って持続的な検証の対象となっていること。 |
(続き) エンドトキシン/発熱性物質、微粒子及び微生物の汚染の外因性発生源となり得るもの(人員、原材料及び周囲環境等)から製品の保護を高めるとともに、環境及び製品中にある汚染物質となり得るものの迅速な検出に資するように、適切な技術(例:アクセス制限バリアシステム(RABS)、アイソレータ、ロボットシステム、迅速・代替法及び連続モニタリングシステム)の利用が検討されていること。 |
4 建物 4.1 無菌製品の製造が適切なクリーンルーム内で行われていること、当該クリーンルームには、エアロックとして機能する更衣室を通って人員が入室し、且つ設備及び原材料がエアロックを通して搬入されていること。クリーンルーム及び更衣室は、適切な清浄度基準に維持管理され、且つ適切な効率のフィルタを通った空気が供給されていること。管理及び監視では、科学的に妥当性を示すこと、且つクリーンルーム、エアロック及びパススルーハッチについての環境条件の状態を効果的に評価すること。 |
4.15 クリーンルーム及び清浄区画内での気流パターンを視覚化して、低い等級から高い等級の区域へ流入することがない旨、また、低い等級の区域(床など) から又はより高い等級の区域へ汚染を運ぶおそれのある作業者又は設備の上を空気が漂うことのない旨を、実証すること。 (略) 空気の視覚化検討試験の結果を文書化するとともに、その施設の環境モニタリングプログラムを策定する際に検討すること。 |
クリーンルーム及び清浄空気設備の適格性評価 4.24 クリーンルーム及び清浄空気設備は,アネックス15の要求事項に準拠する方法を用いて適格性評価されていること。クリーンルームの適格性評価(等級分けを含む) は、運用時の環境モニタリングと明確に区別されていること。 |
5 設備 5.9 微粒子計数器(サンプリング管を含む)は、適格性評価されていること。チューブの直径及び曲がり半径については、その製造元の推奨仕様を検討すること。チューブの長さは一般的に1m以内であること( なお、妥当性が示されているときには、この限りでない) 、且つ屈曲箇所の数が最小化されていること。サンプリング管の長さが短い携帯式微粒子計数器は、等級分け目的で用いること。等速サンプリングヘッドは、一方向気流システムにおいて用いること。微粒子計数器は、適切な方向に向けて重要箇所にできるだけ近く配置して、採取検体が全体を反映するものであることを確保すること。 |
9 環境及び工程のモニタリング 全般事項 9.1 その製造所の環境及び工程のモニタリングプログラムは、そのCCS全体の一部を形成するとともに、微生物・微粒子汚染のリスクを最小化するように設計された管理をモニターするため用いられる。そのモニタリングシステムの各要素(生菌、非生菌及びAPS)を単独で捉えてもその信頼性は限定的であって、個別に無菌状態の指標とみなしてはならないことに留意すること。それらの結果は、併せて検討するときに、それらがモニターしているシステムの設計、バリデーション及び運用の信頼性を確認することに役立つ。 9.2 本プログラムは一般的に、以下の要素で構成される: i . 環境モニタリングー 総微粒子量 |
環境及び工程のモニタリング 9.4 環境モニタリングプログラムが確立され、文書化されていること。環境モニタリングプログラムの目的は以下のとおり: i . クリーンルーム及び清浄空気設備が、設計及び規制上の要求事項に従って、適切な空気清浄度の環境を提供し続けることの保証を得る。 ii. 環境限度値からの外れ値を効果的に検知して、原因調査及び製品品質へのリスクの評価の端緒とする。 |
(続き) こうした包括的な環境モニタリングプログラム、即ち検体採取箇所、モニタリングの頻度、モニタリング方法及び培養条件(例: 時間、温度、好気性及び/又は嫌気性条件)を確立するために、リスク評価を行うこと。 |
(続き) これらのリスク評価は、以下の詳細な知識に基づいて実施すること;工程でのインプット及び最終的な製品、その施設、設備、特定の工程及びステップの重要度、関連する作業、通常時のモニタリングデータ、適格性評価の際に得られたモニタリングデータ、及び環境から分離される典型的な微生物叢についての知識。 |
(続き) そのリスク評価には、重要なモニタリング箇所、即ち工程処理の際に微生物が存在することで製品品質へのインパクトを有し得る箇所(例:グレードAの無菌操作区域、及びグレードA区域と直接連結するグレードB 区域)の決定を含めること。空気視覚化検討試験等、その他の情報の検討も含まれていること。これらのリスク評価は、その製造所の環境モニタリングプログラムの有効性を確認するため、定期的に照査されていること。そのモニタリングプログラムは、その製造所の傾向分析及びCCSの全体的な状況において、検討されるものであること。 |
9.5 クリーンルーム、清浄空気設備及び人 員について通常時のモニタリングが、工程の重要段階全て(設備の始動準備を含む) に亘って作業中に行われていること。 9.7 グレードAについてのモニタリング は、重要作業の際に無菌操作条件が保たれていることを実証するものであること。無菌設備表面、容器、密栓及び製品への汚染のリスクが最も高くなる箇所で、モニタリングを行うこと。モニタリング箇所の選定並びに検体採取装置の向き及び配置は、妥当性が示されていて、且つ重要区域から信頼できるデータを得るのに適切なものであること。 9.8 検体採取方法が、製造作業に汚染のリスクをもたらしてはならない。 |
環境モニタリング ー 総微粒子量 9.15 グレード毎の浮遊微粒子汚染の環境モニタリングの限度値を、表5に示す |
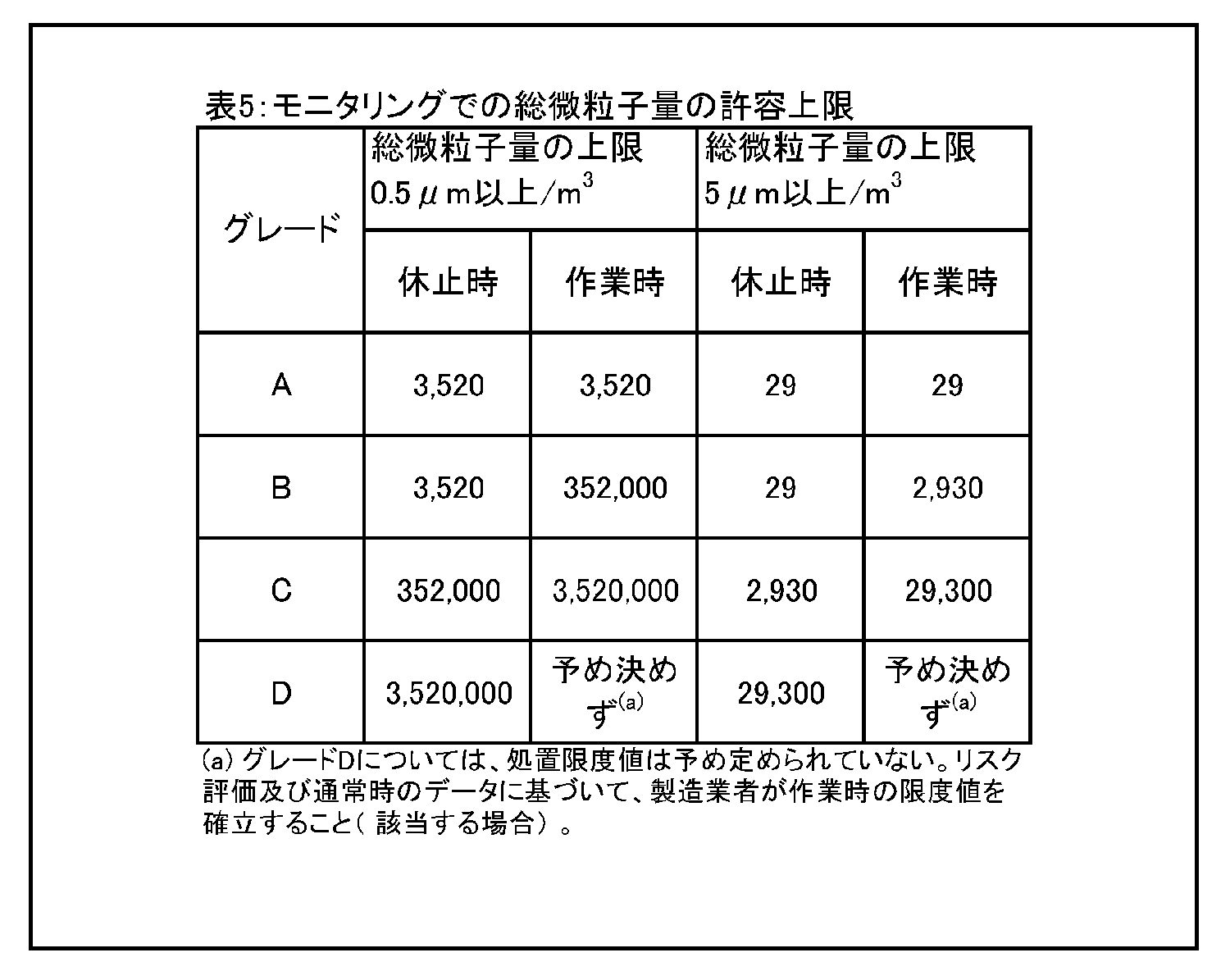 |
注1: 「非作業時」状態について表中に示されている微粒子限度値は、作業の完了後に無人状態で、適格性評価の際に定められた短時間の「クリーンアップ」期(ガイダンス値として20分未満) を経た後に達成されるものであること(4.29節を参照) 。 注2:グレードA中においてマクロ粒子( 特に5μm以上のもの) 計数が時折表示される場合は、電気的なノイズ、迷光、偶然一致した減損等による誤計数であると考えてよい。ただし、低レベルで連続した又は規則的な計数があるときには、汚染事象の可能性を示唆していることがあり得るので調査すること。そうした事象は、室内空気供給のフィルタ処理システムの初期不具合、設備不具合を示しているおそれがあり、又は機械の始動準備及び通常時の作業の際の杜撰な慣行を診断することにもなり得る。 |
9.16 グレードAについては、重要操作( 設備の部品組立てを含む) の間は常時粒子のモニタリングがなされること。 9.17 グレードA区域は、(0.5μm以上及び5μm以上の微粒子について) 連続的に、且つ適切な検体流量( 少なくとも毎分2 8 リッター(1ft3) ) でモニターして、全ての介入操作、一過性の事象及びシステム低下が捕捉されるようにすること。そのシステムは、各個別検体の結果について、潜在的な外れ値を同定して適時に対応することができるような頻度で、警報基準値及び処置限度値との相関を頻繁に示すものであること。警報基準値を超過しているときには、警報が発せられること。追加の微生物モニタリングの検討を含めて、警報に対応してとるべき処置が、手順に定められていること。 |
9.18 検体採取の頻度は減らし得るが、グレードB区域に同様のシステムを用いることが推奨される。グレードB区域は、汚染のレベルの増大及びシステム低下があれば、そのプログラムが捕捉することができるような頻度及び適切な検体サイズで、モニタリングすること。警報基準値を超過しているときには、警報が発せられること。 9.21 自動化システムを用いて採取されるモニタリング検体のサイズは、通常、当該システムの検体採取速度との相関関係になる。検体量について、クリーンルーム及び清浄空気設備の正式な等級分けに用いられた量と同じである必要はない。モニタリング検体量には、妥当性が示されていること。 |
用語解説 汚染制御ストラテジー(CCS) - 微生物、エンドトキシン/発熱性物質及び微粒子を制御する計画一式であり、現行の製品及び工程の理解から導き出され、工程性能及び製品品質を保証するもの。当該制御には、原薬、添加剤並びに薬剤の原材料及び構成物、施設及び設備の操作条件、工程内管理、最終製品の規格、並びに付随するモニタリング及び管理の頻度及び方法に関連するパラメータ及び特性が含まれ得る。 |
用語解説 グレードA空気供給 - 総微粒子量がグレードA品質の空気を産することができるものとして適格性評価されたフィルタを通した空気(なお、持続的に総微粒子量モニタリングを行う要求事項はなく、グレードAの生菌モニタリング限度値に合致する要求事項もない)。キャップがまだ巻締めされていないが完全に止栓されているバイアルの保護に特に用いられる。 |
用語解説 エアロック - インターロック付きドアのある囲まれた空間で、隣接する室(一般に、異なる空気清浄基準が異なるもの)間の気圧制御を保持するよう構築されたもの。エアロックの目的は、より低い管理がなされる区域から微粒子物質及び微生物の汚染が入り込むのを防ぐことである。 |
用語解説 パススルーハッチ - エアロック(エアロックの定義を参照) と同義であるが、一般的に寸法が小さいもの。 |
パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.11 閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセス( 例: 添加剤、培地、緩衝液、ガスを添加する間のプロセス) では、適切な環境条件を適用すること。無菌操作については、アネックス1 に準拠したパラメータ( すなわち、グレードB を背景環境とするグレードA ) を適用すること。環境モニタリングのプログラムには、非微生物汚染、微生物汚染及び空気差圧の試験及びモニタリングを含めること。モニターする場所は、Q R M の原則を考慮して決定すること。モニタリングの検体数、量及び頻度並びにアラート及び対処の限度値は、Q R M の原則を考慮して適切であること。 |
3.13 閉鎖システムについて、Q R M 評価の結果に基づいて、グレードB の背景環境におけるグレードA より低い清浄度区分の区域が許容される場合があり得る。空気の清浄度区分とモニタリングの適切なレベルは、当該製品の性質、製造工程及び使用する設備を検討し、具体的なリスクを考慮して決定すること。 | IV. BUILDINGS AND FACILITIES A. Critical Area - Class 100 (ISO 5) (略)We recommend conducting nonviable particle monitoring with a remote counting system. These systems are capable of collecting more comprehensive data and are generally less invasive than portable particle counters. See Section X.E. for additional guidance on particle monitoring. |
APPENDIX 3: PROCESSING PRIOR TO FILLING AND SEALING OPERATIONS A. Aseptic processing from early manufacturing steps (略)Procedures (e.g., aseptic connection) that expose a product or product contact surfaces should be performed under unidirectional airflow in a Class 100 (ISO 5H9:K9) environment. The environment of the room surrounding the Class 100 (ISO 5) environment should be Class 10,000 (ISO 7) or better. Microbiological and airborne particle monitoring should be performed during operations. Microbial surface monitoring should be performed at the end of operations, but prior to cleaning. vi. Controlled removal of dust close to source of the contaminant e.g. through localised extraction; |
Chapter 5: Production Prevention of cross-contamination in production 5.21 The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organisational measures required to control risks for cross-contamination. These could include, but are not limited to, the following: Organisational Measures iv. Depending on the contamination risk, verification of cleaning of non product contact surfaces and monitoring of air within the manufacturing area and/or adjoining areas in order to demonstrate effectiveness of control measures against airborne contamination or contamination by mechanical transfer; |
2 Principle 2.1 The manufacture of sterile products is subject to special requirements in order to minimize risks of microbial, particulate and endotoxin/pyrogen contamination. The following key areas should be considered: i.Facility, equipment and process should be appropriately designed, qualified and/or validated and where applicable, subjected to ongoing verification according to the relevant sections of the Good Manufacturing Practices (GMP) guidelines. |
(続き) The use of appropriate technologies (e.g. Restricted Access Barriers Systems (RABS), isolators, robotic systems, rapid/alternative methods and continuous monitoring systems) should be considered to increase the protection of the product from potential extraneous sources of endotoxin/pyrogen, particulate and microbial contamination such as personnel, materials and the surrounding environment, and assist in the rapid detection of potential contaminants in the environment and the product. |
4 Premises 4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to which should be through change rooms that act as airlocks for personnel and airlocks for equipment and materials. Cleanrooms and change rooms should be maintained to an appropriate cleanliness standard and supplied with air that has passed through filters of an appropriate efficiency. Controls and monitoring should be scientifically justified and should effectively evaluate the state of environmental conditions of cleanrooms, airlocks and pass-through hatches. |
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is no ingress from lower grade to higher grade areas and that air does not travel from less clean areas (such as the floor) or over operators or equipment that may transfer contamination to the higher grade areas.(中略) The outcome of the air visualisation studies should be documented and considered when establishing the facility's environmental monitoring programme. | Cleanroom and clean air equipment qualification 4.24 Cleanrooms and clean air equipment should be qualified using methodology in accordance with the requirements of Annex 15. Cleanroom qualification (including classification) should be clearly differentiated from operational environmental monitoring. | 5 Equipment 5.9 Particle counters, including sampling tubing, should be qualified. The manufacturer’s recommended specifications should be considered for tube diameter and bend radii. Tube length should typically be no longer than 1m unless justified and the number of bends should be minimized. Portable particle counters with a short length of sample tubing should be used for classification purposes. Isokinetic sampling heads should be used in unidirectional airflow systems. They should be oriented appropriately and positioned as close as possible to the critical location to ensure that samples are representative. |
9 Environmental & process monitoring General 9.1 The site’s environmental and process monitoring programme forms part of the overall CCS and is used to monitor the controls designed to minimize the risk of microbial and particle contamination. It should be noted that the reliability of each of the elements of the monitoring system (viable, non-viable and APS) when taken in isolation is limited and should not be considered individually to be an indicator of asepsis. 9.2 This programme is typically comprised of the following elements: i. Environmental monitoring - total particle. |
Environmental and process monitoring 9.4 An environmental monitoring programme should be established and documented. The purpose of the environmental monitoring programme, is to: i. Provide assurance that cleanrooms and clean air equipment continue to provide an environment of appropriate air cleanliness, in accordance with design and regulatory requirements. ii. Effectively detect excursions from environmental limits triggering investigation and assessment of risk to product quality. |
(続き) Risk assessments should be performed in order to establish this comprehensive environmental monitoring programme, i.e. sampling locations, frequency of monitoring, monitoring methods and incubation conditions (e.g. time, temperature(s), aerobic and/or anaerobic conditions). These risk assessments should be conducted based on detailed knowledge of; the process inputs and final product, the facility, equipment, the criticality of specific processes and steps, the operations involved, routine monitoring data, monitoring data obtained during qualification and knowledge of typical microbial flora isolated from the environment. |
(続き) The risk assessment should include the determination of critical monitoring locations, those locations where the presence of microorganisms during processing may have an impact upon product quality, (e.g. grade A, aseptic processing areas and the grade B areas that directly interface with the grade A area). Consideration of other information such as air visualisation studies should also be included. These risk assessments should be reviewed regularly in order to confirm the effectiveness of the site’s environmental monitoring programme. The monitoring programme should be considered in the overall context of the trend analysis and the CCS for the site. |
9.5 Routine monitoring of cleanrooms, clean air equipment and personnel should be performed in operation throughout all critical stages of processing, including equipment set-up. 9.7 The monitoring of grade A should demonstrate the maintenance of aseptic processing conditions during critical operations. Monitoring should be performed at locations posing the highest risk of contamination to the sterile equipment surfaces, containers, closures and product. The selection of monitoring locations and the orientation and positioning of sampling devices should be justified and appropriate to obtain reliable data from the critical zones. 9.8 Sampling methods should not pose a risk of contamination to the manufacturing operations. |
Environmental monitoring - Total Particle 9.15 The limits for environmental monitoring of airborne particle concentration for each graded area are given in Table 5. |
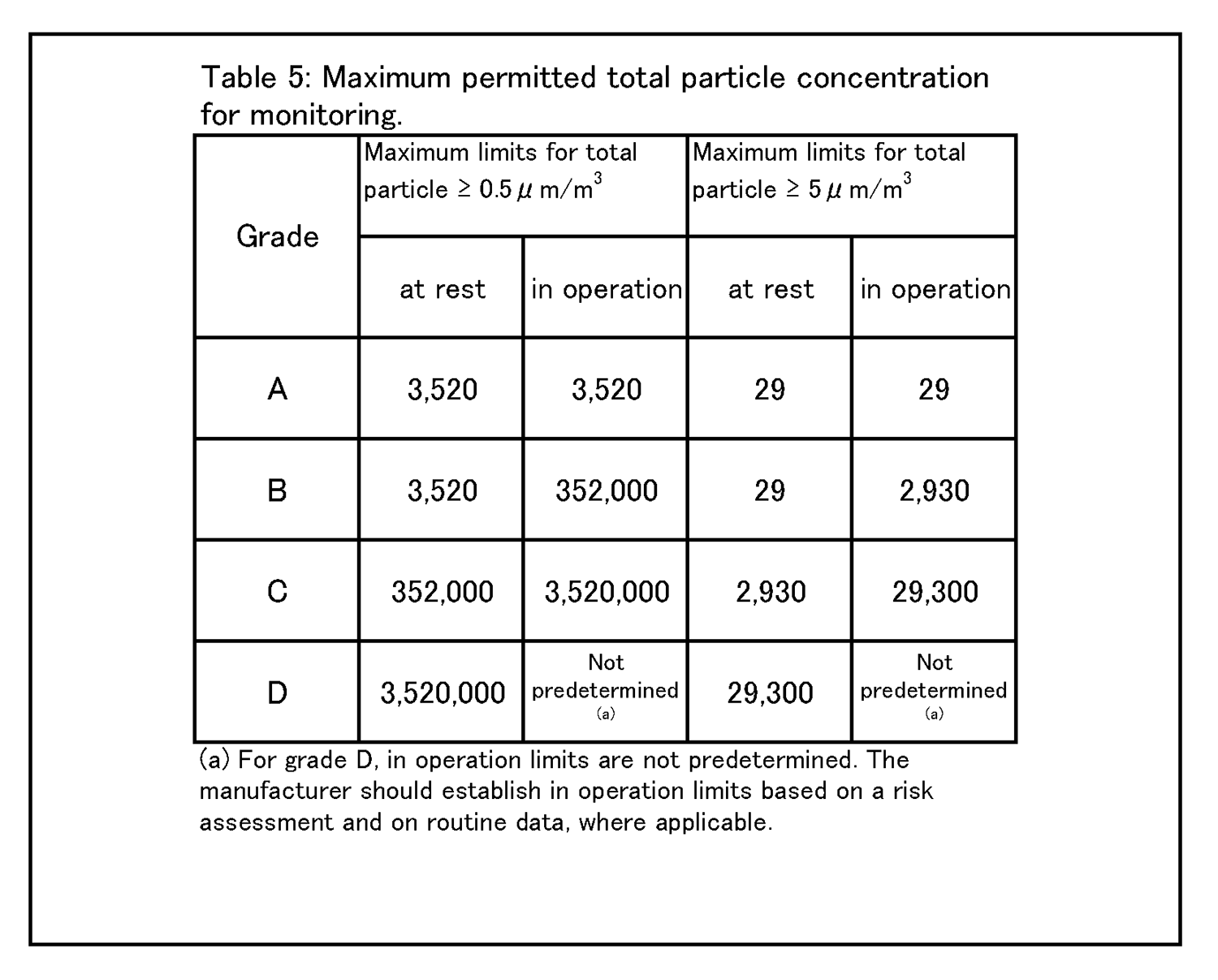 |
Note 1:The particle limits given in the table for the “at rest” state should be achieved after a short “clean up” period defined during qualification (guidance value of less than 20 minutes) in an unmanned state, after the completion of operations (see paragraph 4.29). Note 2:The occasional indication of macro particle counts, especially ≧ 5 μm, within grade A may be considered to be false counts due to electronic noise, stray light, coincidence loss etc. However, consecutive or regular counting of low levels may be indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the room air supply filtration system, equipment failure, or may also be diagnostic of poor practices during machine set-up and routine operation. |
9.16 For grade A, particle monitoring should be undertaken for the full duration of critical processing, including equipment assembly. 9.17 The grade A area should be monitored continuously (for particles ≧0.5 and ≧5 μm) and with a suitable sample flow rate (at least 28 litres (1ft3) per minute) so that all interventions, transient events and any system deterioration is captured. The system should frequently correlate each individual sample result with alert levels and action limits at such a frequency that any potential excursion can be identified and responded to in a timely manner. Alarms should be triggered if alert levels are exceeded. Procedures should define the actions to be taken in response to alarms including the consideration of additional microbial monitoring. |
9.18 It is recommended that a similar system be used for the grade B area although the sample frequency may be decreased. The grade B area should be monitored at such a frequency and with suitable sample size that the programme captures any increase in levels of contamination and system deterioration. If alert levels are exceeded, alarms should be triggered. 9.21 The size of monitoring samples taken using automated systems will usually be a function of the sampling rate of the system used. It is not necessary for the sample volume to be the same as that used for formal classification of cleanrooms and clean air equipment. Monitoring sample volumes should be justified. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Glossary Grade A air supply - Air which is passed through a filter qualified as capable of producing grade A total particle quality air, but where there is no requirement to perform continuous total particle monitoring or meet grade A viable monitoring limits. Specifically used for the protection of fully stoppered vials where the cap has not yet been crimped. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
Chapter 5: Production Prevention of cross-contamination in production 5.21 The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organisational measures required to control risks for cross-contamination. These could include, but are not limited to, the following: Organisational Measures iv. Depending on the contamination risk, verification of cleaning of non product contact surfaces and monitoring of air within the manufacturing area and/or adjoining areas in order to demonstrate effectiveness of control measures against airborne contamination or contamination by mechanical transfer; |
2 Principle 2.1 The manufacture of sterile products is subject to special requirements in order to minimize risks of microbial, particulate and endotoxin/pyrogen contamination. The following key areas should be considered: i.Facility, equipment and process should be appropriately designed, qualified and/or validated and where applicable, subjected to ongoing verification according to the relevant sections of the Good Manufacturing Practices (GMP) guidelines. |
(続き) The use of appropriate technologies (e.g. Restricted Access Barriers Systems (RABS), isolators, robotic systems, rapid/alternative methods and continuous monitoring systems) should be considered to increase the protection of the product from potential extraneous sources of endotoxin/pyrogen, particulate and microbial contamination such as personnel, materials and the surrounding environment, and assist in the rapid detection of potential contaminants in the environment and the product. |
4 Premises 4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to which should be through change rooms that act as airlocks for personnel and airlocks for equipment and materials. Cleanrooms and change rooms should be maintained to an appropriate cleanliness standard and supplied with air that has passed through filters of an appropriate efficiency. Controls and monitoring should be scientifically justified and should effectively evaluate the state of environmental conditions of cleanrooms, airlocks and pass-through hatches. |
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is no ingress from lower grade to higher grade areas and that air does not travel from less clean areas (such as the floor) or over operators or equipment that may transfer contamination to the higher grade areas.(中略) The outcome of the air visualisation studies should be documented and considered when establishing the facility's environmental monitoring programme. | Cleanroom and clean air equipment qualification 4.24 Cleanrooms and clean air equipment should be qualified using methodology in accordance with the requirements of Annex 15. Cleanroom qualification (including classification) should be clearly differentiated from operational environmental monitoring. | 5 Equipment 5.9 Particle counters, including sampling tubing, should be qualified. The manufacturer’s recommended specifications should be considered for tube diameter and bend radii. Tube length should typically be no longer than 1m unless justified and the number of bends should be minimized. Portable particle counters with a short length of sample tubing should be used for classification purposes. Isokinetic sampling heads should be used in unidirectional airflow systems. They should be oriented appropriately and positioned as close as possible to the critical location to ensure that samples are representative. |
9 Environmental & process monitoring General 9.1 The site’s environmental and process monitoring programme forms part of the overall CCS and is used to monitor the controls designed to minimize the risk of microbial and particle contamination. It should be noted that the reliability of each of the elements of the monitoring system (viable, non-viable and APS) when taken in isolation is limited and should not be considered individually to be an indicator of asepsis. 9.2 This programme is typically comprised of the following elements: i. Environmental monitoring - total particle. |
Environmental and process monitoring 9.4 An environmental monitoring programme should be established and documented. The purpose of the environmental monitoring programme, is to: i. Provide assurance that cleanrooms and clean air equipment continue to provide an environment of appropriate air cleanliness, in accordance with design and regulatory requirements. ii. Effectively detect excursions from environmental limits triggering investigation and assessment of risk to product quality. |
(続き) Risk assessments should be performed in order to establish this comprehensive environmental monitoring programme, i.e. sampling locations, frequency of monitoring, monitoring methods and incubation conditions (e.g. time, temperature(s), aerobic and/or anaerobic conditions). These risk assessments should be conducted based on detailed knowledge of; the process inputs and final product, the facility, equipment, the criticality of specific processes and steps, the operations involved, routine monitoring data, monitoring data obtained during qualification and knowledge of typical microbial flora isolated from the environment. |
(続き) The risk assessment should include the determination of critical monitoring locations, those locations where the presence of microorganisms during processing may have an impact upon product quality, (e.g. grade A, aseptic processing areas and the grade B areas that directly interface with the grade A area). Consideration of other information such as air visualisation studies should also be included. These risk assessments should be reviewed regularly in order to confirm the effectiveness of the site’s environmental monitoring programme. The monitoring programme should be considered in the overall context of the trend analysis and the CCS for the site. |
9.5 Routine monitoring of cleanrooms, clean air equipment and personnel should be performed in operation throughout all critical stages of processing, including equipment set-up. 9.7 The monitoring of grade A should demonstrate the maintenance of aseptic processing conditions during critical operations. Monitoring should be performed at locations posing the highest risk of contamination to the sterile equipment surfaces, containers, closures and product. The selection of monitoring locations and the orientation and positioning of sampling devices should be justified and appropriate to obtain reliable data from the critical zones. 9.8 Sampling methods should not pose a risk of contamination to the manufacturing operations. |
9.15 The limits for environmental monitoring of airborne particle concentration for each graded area are given in Table 5. |
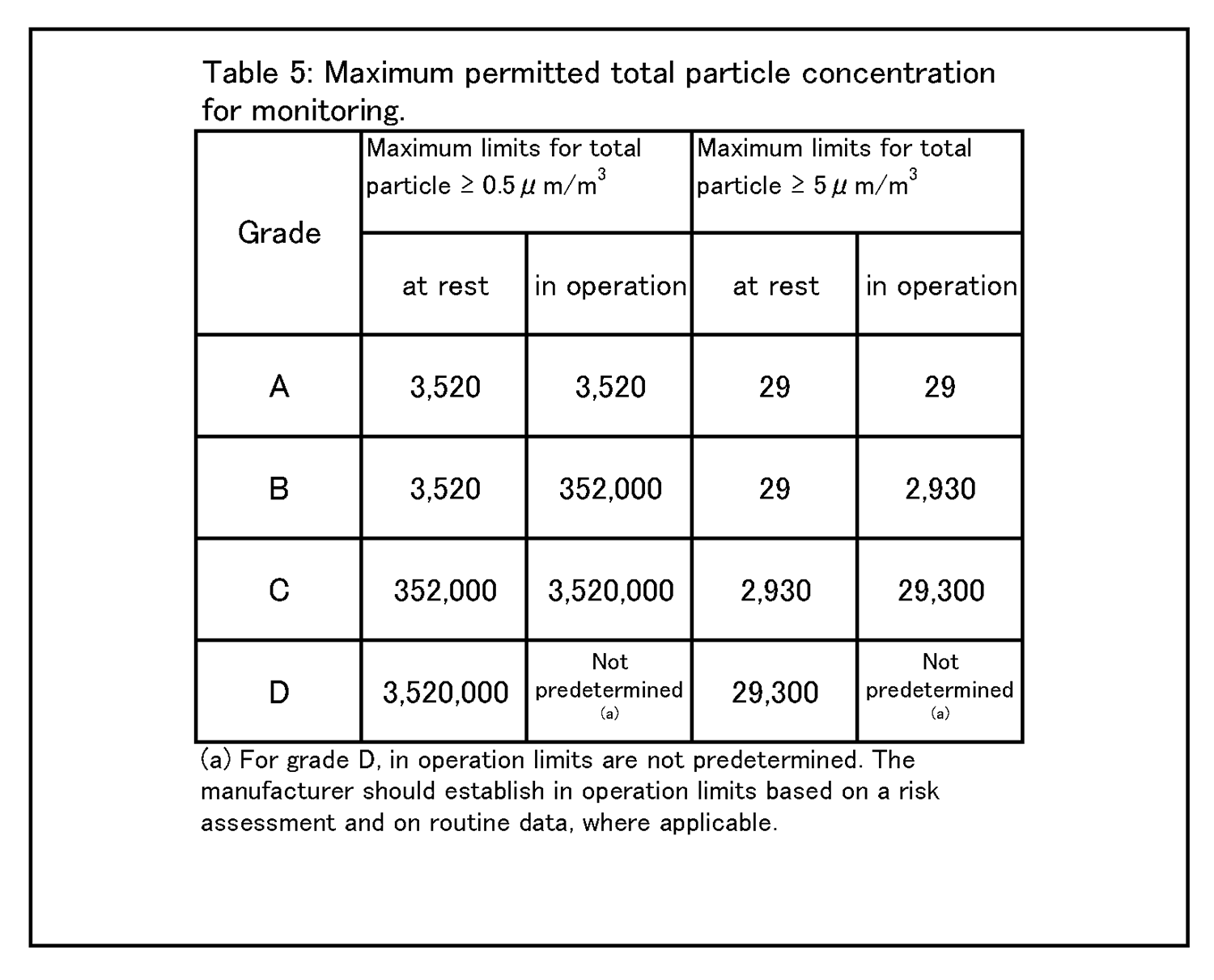 |
Note 1:The particle limits given in the table for the “at rest” state should be achieved after a short “clean up” period defined during qualification (guidance value of less than 20 minutes) in an unmanned state, after the completion of operations (see paragraph 4.29). Note 2:The occasional indication of macro particle counts, especially ≧ 5 μm, within grade A may be considered to be false counts due to electronic noise, stray light, coincidence loss etc. However, consecutive or regular counting of low levels may be indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the room air supply filtration system, equipment failure, or may also be diagnostic of poor practices during machine set-up and routine operation. |
9.16 For grade A, particle monitoring should be undertaken for the full duration of critical processing, including equipment assembly. 9.17 The grade A area should be monitored continuously (for particles ≧0.5 and ≧5 μm) and with a suitable sample flow rate (at least 28 litres (1ft3) per minute) so that all interventions, transient events and any system deterioration is captured. The system should frequently correlate each individual sample result with alert levels and action limits at such a frequency that any potential excursion can be identified and responded to in a timely manner. Alarms should be triggered if alert levels are exceeded. Procedures should define the actions to be taken in response to alarms including the consideration of additional microbial monitoring. |
9.18 It is recommended that a similar system be used for the grade B area although the sample frequency may be decreased. The grade B area should be monitored at such a frequency and with suitable sample size that the programme captures any increase in levels of contamination and system deterioration. If alert levels are exceeded, alarms should be triggered. 9.21 The size of monitoring samples taken using automated systems will usually be a function of the sampling rate of the system used. It is not necessary for the sample volume to be the same as that used for formal classification of cleanrooms and clean air equipment. Monitoring sample volumes should be justified. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Glossary Grade A air supply - Air which is passed through a filter qualified as capable of producing grade A total particle quality air, but where there is no requirement to perform continuous total particle monitoring or meet grade A viable monitoring limits. Specifically used for the protection of fully stoppered vials where the cap has not yet been crimped. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
2. Principle 2.1 The manufacture of sterile products is subject to specific requirements in order to minimize risks of microbial, particulate and endotoxin/pyrogen contamination. As a minimum, the following areas should be considered: i. Premises, equipment and process should be appropriately designed, qualified and validated and, where applicable, be subjected to ongoing verification according to the relevant sections of the good manufacturing practices (GMP) guide. |
(続き) The use of appropriate technologies (such as restricted access barrier systems (RABS), isolators, robotic systems, rapid/alternative methods and continuous monitoring systems) should be considered to increase the protection of the product from potential sources of endotoxin/pyrogen, particulate and microbial contamination, such as personnel, materials and the surrounding environment, and assist in the rapid detection of potential contaminants in the environment and the product. |
4. Premises 4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to which should be through change rooms that act as airlocks. Cleanrooms and change rooms should be maintained at an appropriate cleanliness standard and supplied with air that has passed through filters of an appropriate efficiency. Controls and monitoring should be scientifically justified and should effectively evaluate the state of environmental conditions of cleanrooms, airlocks and pass-through hatches. |
4.15 Airflow pattern studies should be performed both at rest and in operation (for example, simulating operator interventions). Video recordings of the airflow patterns should be carried out by following good practices to demonstrate the above. Recordings should be retained. The outcome of the air visualization studies should be documented and taken into consideration when establishing the facility’s environmental monitoring programme. | Cleanroom and clean air equipment qualification 4.24 Cleanrooms and clean air equipment should be qualified using methodology in accordance with the requirements of the WHO Good manufacturing practices: guideline on validation.3 Cleanroom qualification (including classification) should be clearly differentiated from operational environmental monitoring. | 5. Equipment 5.9 Particle counters, including sampling tubing, should be qualified. The manufacturer’s recommended specifications should be considered for tube diameter and bend radii. Tube length should typically be no longer than 1 m unless justified, and the number of bends should be minimized. Portable particle counters with a short length of sample tubing should be used for classification purposes. Isokinetic sampling heads should be used in unidirectional airflow systems. They should be oriented appropriately and positioned as close as possible to the critical location to ensure that samples are representative. |
9. Environmental and process monitoring General 9.1 The site’s environmental and process monitoring programme forms part of the overall CCS and is used to monitor the controls designed to minimize the risk of microbial and particle contamination. It should be noted that the reliability of each of the elements of the monitoring system (viable, non-viable and APS) when taken in isolation is limited and should not be considered individually to be an indicator of asepsis. 9.2 This programme typically comprises the following elements: i. environmental monitoring - total particle |
Environmental and process monitoring 9.4 An environmental monitoring programme should be established and documented. The purpose of the environmental monitoring programme is to: i. provide assurance that cleanrooms and clean air equipment continue to provide an environment of appropriate air cleanliness, in accordance with design and regulatory requirements; ii. effectively detect excursions from environmental limits triggering investigation and assessment of risk to product quality. |
(続き) Risk assessments should be performed in order to establish this comprehensive environmental monitoring programme, such as sampling locations, frequency of monitoring, monitoring methods and incubation conditions (such as time, temperature, and aerobic or anaerobic conditions). These risk assessments should be conducted based on detailed knowledge of the process inputs and final product, the facility, equipment, the criticality of specific processes and steps, the operations involved, routine monitoring data, monitoring data obtained during qualification and knowledge of typical microbial flora isolated from the environment. |
(続き) The risk assessment should include the determination of critical monitoring locations - those locations where the presence of microorganisms during processing may have an impact upon product quality (for example, grade A aseptic processing areas and grade B areas that directly interface with grade A areas). Consideration of other information, such as air visualization studies, should also be included. These risk assessments should be reviewed regularly in order to confirm the effectiveness of the site’s environmental monitoring programme. The monitoring programme should be considered in the overall context of the trend analysis and the CCS for the site. |
9.5 The routine monitoring of cleanrooms, clean air equipment and personnel should be performed in operation throughout all critical stages of processing, including equipment set-up. 9.7 The monitoring of grade A should demonstrate the maintenance of aseptic processing conditions during critical operations. Monitoring should be performed at locations posing the highest risk of contamination of the sterile equipment surfaces, containers, closures and product. The selection of monitoring locations and the orientation and positioning of sampling devices should be justified and appropriate to obtain reliable data from the critical zones. 9.8 Sampling methods should not pose a risk of contamination of the manufacturing operations. |
9.15 The limits for environmental monitoring of airborne particle concentration for each graded area are given in Table 5. |
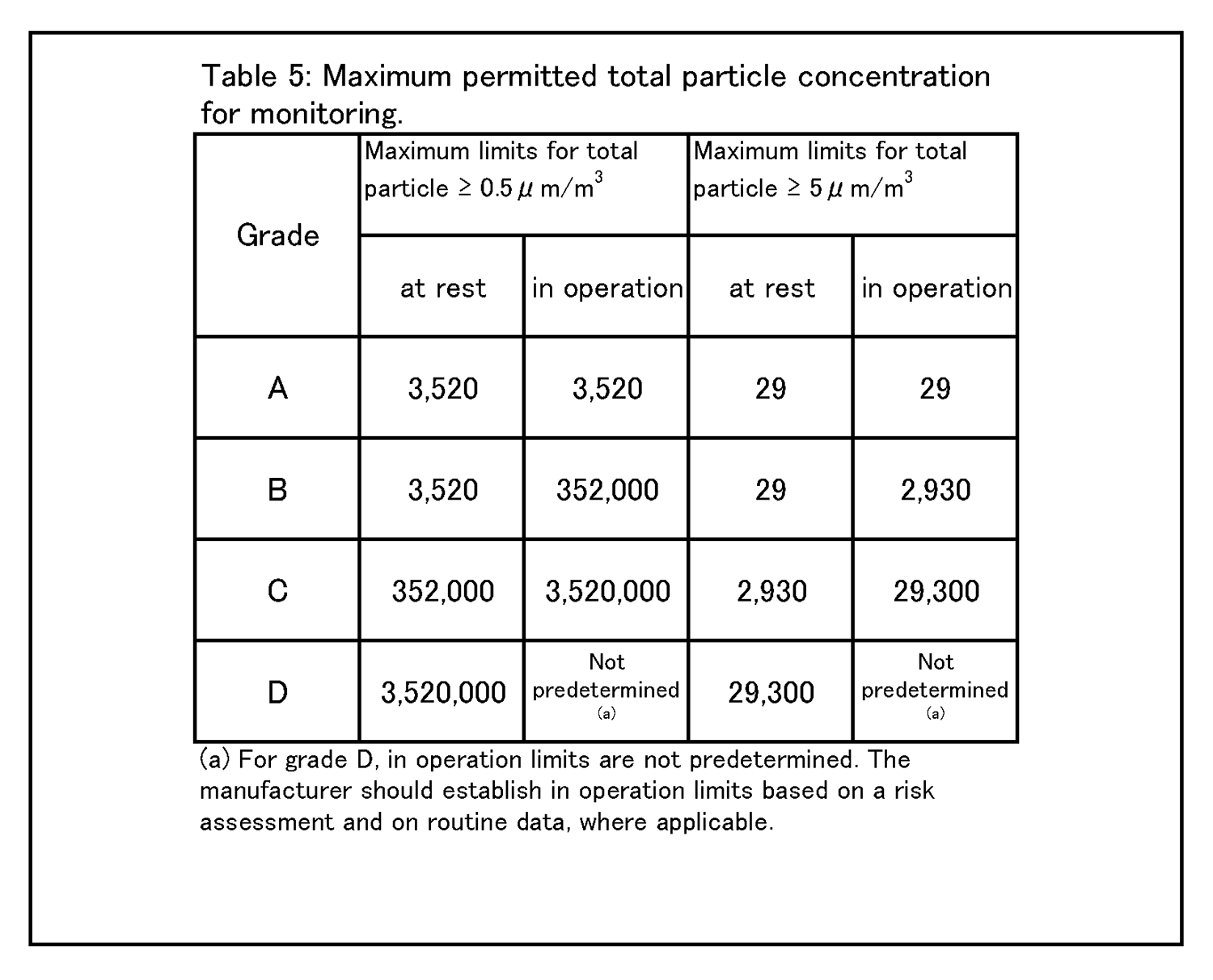 |
Note 1: The particle limits given in the table for the at rest state should be achieved after a short clean-up period defined during qualification (guidance value of less than 20 minutes) in an unmanned state, after the completion of operations (refer to paragraph 4.29). Note 2: The occasional indication of macro particle counts, especially ≧ 5 μm, within grade A may be considered to be false counts due to electronic noise, stray light, coincidence loss, or other factor. However, consecutive or regular counting of low levels may be indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the room air supply filtration system or equipment failure, or may be diagnostic of poor practices during machine set-up and routine operation. |
9.16 For grade A, particle monitoring should be undertaken for the full duration of critical processing, including equipment assembly. 9.17 The grade A area should be monitored continuously (for particles ≧ 0.5 and ≧ 5 μm) and with a suitable sample flow rate (at least 28 litres per minute) so that all interventions,transient events and any system deterioration is captured. The system should frequently correlate each individual sample result with alert levels and action limits at such a frequency that any potential excursion can be identified and responded to in a imely manner. Alarms should be triggered if alert levels are exceeded. Procedures should define the actions to be taken in response to alarms, including the consideration of additional microbial monitoring. |
9.18 It is recommended that a similar system be used for the grade B area, though the sampling frequency may be decreased. The grade B area should be monitored at such a frequency and with suitable sample size that the programme captures any increase in levels of contamination and system deterioration. If alert limits are exceeded, alarms should be triggered. 9.21 The size of monitoring samples taken using automated systems will usually be a function of the sampling rate of the system used. It is not necessary for the sample volume to be the same as that used for formal classification of cleanrooms and clean air equipment. Monitoring sample volumes should be justified. |
Glossary contamination control strategy (CCS). A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Glossary grade A air supply. Air that is passed through a filter qualified as capable of producing grade A total particle quality air, but where there is no requirement to perform continuous total particle monitoring or meet grade A viable monitoring |
Glossary airlock. An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a less controlled area. |
Glossary pass-through hatch. Synonymous with airlock [refer to airlock definition] but typically smaller in size. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・重要区域(グレードA)、直接支援区域(グレードB)の微粒子・微生物数のモニタリングの方法・頻度について。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・浮遊微粒子モニタリングの対象粒径および微粒子管理の連続モニタリング。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・対象粒径、モニタリングポイント、サンプリング量について。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・連続モニタリング、非製造時のモニタリング。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:全般 以下の要件があります。 ・汚染リスクに応じて製造区域及び/ 又は隣接区域内における浮遊微粒子モニタリングを行うこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌製品の製造の汚染の汚染のリスクを最小化するための要求事項としての連続モニタリングシステム。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・環境にある汚染物質を迅速に検出する適切な技術としての連続モニタリングシステム。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルーム、エアロック、パススルーハッチの清浄度モニタリングの科学的な妥当性について。 *用語ついての補足:「エアロック」は本表アネックス1の用語解説を参照ください。 *用語ついての補足:「パススルーハッチ」は本表アネックス1の用語解説を参照ください。 「パスボックス」が「パススルーハッチ」に該当します。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・環境モニタリングプログラムを規定するうえで気流の可視化試験の結果を考慮することついて。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームと空調は適格性評価が必要で、運用時環境モニタリングと区別すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微粒子計数器(サンプリング管を含む)のチューブの直径及び曲がり半径について。 ・微粒子計数器(サンプリング管を含む)のチューブの長さは一般的に1m以内であること。 ・屈曲個所数が最小化されていること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・環境モニタリングについて、汚染管理戦略(CCS)、汚染のリスクを最小化する設計に関連づけた言及があります。 ・環境モニタリングは総微粒子量の要素で構成されます。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・環境モニタリングの目的について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・リスク評価を行い、環境モニタリングプログラムを確立すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・環境モニタリングプログラムに関係したリスク評価について工程や施設に関する詳細な知識に基づいて実施すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・環境モニタリングプログラムに関係したリスク評価についてモニタリング箇所や空気視覚化の検討・モニタリングプログラムの有効性・定期的な見直しを含むこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードAのモニタリングについて ・モニタリングにおけるサンプリング手法について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微粒子量のモニタリングに対する警報基準値及び処置限度値について。 ・モニタリングでの総微粒子量の許容上限。 ・グレードAの計測について。 【誤記訂正】厚生労働省通知の和訳では表5 の最右列、対象粒径5μのグレードB 作業時について""2 9300""と記載されていますが、PIC/Sの原文は""2 930""であるため、誤記です。表は正しい値 2,930を記載。 |
対象医薬品:無菌医薬品 以下の要件があります ・グレードAのモニタリングについて。 【誤記訂正】厚生労働省通知の和訳では「ずっと」と記載されていますが、PIC/Sの原文は full durationであるため、誤記です。記載内容欄(2行目)では「間は常時」を記載。 |
対象医薬品:無菌医薬品 以下の要件があります ・グレードBのモニタリングについて。 ・検体のサイズと量について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCSの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・「グレードA空気供給(Grade A air supply )」として供給する給気においては9.16に示すクリーンルーム等級のグレードA環境と異なり、常時モニタリングは要求されない。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・エアロックの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・パススルーハッチの定義。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセスの微粒子の試験と環境モニタリングについて。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・閉鎖システムにおける清浄度モニタリングのレベル。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・ISO 5の微粒子モニタリングについてその場所に設置する携帯式のパーティクルカウンターより、リモート式の清浄度集中監視システムを推奨。 ・リモート式は、包括的データを収集できること、及びその設置により与える環境負荷が少ない。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・ISO 5の充填・閉塞工程において、微生物及び浮遊微粒子モニタリングを操作中に実施すること。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第5章 製造 製造における交叉汚染の防止 5.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.24と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.9 と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.1、9.2と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.5、9.7、9.8と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリングv 環境モニタリング ー 総微粒子量 9.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 微粒子量 9.16、9.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 微粒子量 9.18、9.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 グレードA空気供給と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第5章 製造 製造における交叉汚染の防止 5.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.24と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.9 と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.1、9.2と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.5、9.7、9.8と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 総微粒子量 9.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 微粒子量 9.16、9.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 微粒子量 9.18、9.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 グレードA空気供給と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.24と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.9 と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 9.1、9.2と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.5、9.7、9.8と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 総微粒子量 9.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 微粒子量 9.16、9.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境モニタリング ー 微粒子量 9.18、9.21と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 グレードA空気供給と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
||||||||
| 国 | 日本 | 日本 | 日本 | 日本 | 米国 | ― | ― | 欧州 | ― | ― | ― | ― | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | PIC/S | EUROPEAN COMMISSION | WHO | WHO | WHO | WHO | ||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3) PIC/S GMPガイドライン アネックス2A ヒト用先端医療医薬品の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing ― Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 2A MANUFACTURE OF ADVANCED THERAPY MEDICINAL PRODUCTS FOR HUMAN USE |
EU Guidelines for Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 54th report Annex 2 International Atomic Energy Agency and World Health Organization guideline on good manufacturing practices for radiopharmaceutical products WHO Technical Report Series, No. 1025, 2020 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||
| 発行年 | 2011年 | 2012年 | 2025年 | 2022年 | 2004年 | 2023年 | 2023年 | 2022年 | 2018年 | 2019年 | 2020年 | 2022年 | ||||||||||
| 記載内容 | 7. 無菌医薬品に係る製品の作業所 7.2 空調システム 7.2.2 空気 3)製品の無菌性を確保する上で特に重要と考えられる差圧については,モニタリングを常時行うこと.さらに,異常時に備えて警報システムを備えることが望ましい。 |
6. 最終滅菌法による医薬品の製造区域 6.2 空調システム 6.2.2 空気 1) 空気の清浄度レベルの異なる区域の間には、室間差圧を設定し、管理及び監視を行うこと。 2) 滅菌前製品のバイオバーデンを管理する上で特に重要と考えられる差圧については、監視測定を常時行うこと。さらに、異常時に備えて警報システムを備えることが望ましい。 3) その他、グレードA/B区域が存在する場合は、原則として無菌操作法による無菌医薬品の製造に関する指針の要件に準じること。 |
4 建物 4.4 無菌製品の製造には、クリーンルーム/ 区画について4つの清浄度等級がある。 グレードB: 無菌操作法による調製・容器充填作業のための、(アイソレータでない場合に)グレードA のバックグラウンドのクリーンルームである。差圧が連続してモニターされていること。 4.16 気圧差の表示器が、クリーンルーム及び/又はアイソレータとそのバックグラウンドとの間に取り付けられていること。設置箇所及び気圧差の重要度が、CCSの中で検討されていること。重要と特定された気圧差は、連続してモニターされ且つ記録作成されていること。 |
(続き) 警報システムが整っていて、空気供給に不具合及び気圧差の低下( 重要と特定された箇所について設定された限度値を下回る) があれば作業者に即時に表示され警報を発するようになっていること。警報シグナルを評価なしに無効としてはならず、警報シグナルが発せられたときにとるべき手立てを概説する手順が用意されていること。警報の遅延を設定する場合には、CCSにおいて評価し、妥当性を示すこと。その他の気圧差は、規則的な間隔でモニターされ且つ記録されていること。 |
用語解説 汚染制御ストラテジー(CCS) - 微生物、エンドトキシン/発熱性物質及び微粒子を制御する計画一式であり、現行の製品及び工程の理解から導き出され、工程性能及び製品品質を保証するもの。当該制御には、原薬、添加剤並びに薬剤の原材料及び構成物、施設及び設備の操作条件、工程内管理、最終製品の規格、並びに付随するモニタリング及び管理の頻度及び方法に関連するパラメータ及び特性が含まれ得る。 |
パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.11 閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセス( 例: 添加剤、培地、緩衝液、ガスを添加する間のプロセス) では、適切な環境条件を適用すること。 無菌操作については、アネックス1 に準拠したパラメータ( すなわち、グレードB を背景環境とするグレードA ) を適用すること。環境モニタリングのプログラムには、非微生物汚染、微生物汚染及び空気差圧の試験及びモニタリングを含めること。モニターする場所は、Q R M の原則を考慮して決定すること。モニタリングの検体数、量及び頻度並びにアラート及び対処の限度値は、Q R M の原則を考慮して適切であること。 |
IV. BUILDINGS AND FACILITIES C. Clean Area Separation The Agency recommends that pressure differentials between cleanrooms be monitored continuously throughout each shift and frequently recorded. All alarms should be documented and deviations from established limits should be investigated. A suitable facility monitoring system will rapidly detect atypical changes that can compromise the facility’s environment. An effective system facilitates restoration of operating conditions to established, qualified levels before reaching action levels. |
(続き) For example, pressure differential specifications should enable prompt detection (i.e., alarms) of an emerging low pressure problem to preclude ingress of unclassified air into a classified room. |
4 Premises 4.4 For the manufacture of sterile products, there are four grades of cleanroom/zone. Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. 4.16 Indicators of air pressure differences should be fitted between cleanrooms and/or between isolators and their background. Set points and the criticality of air pressure differences should be considered within the CCS. Air pressure differences identified as critical should be continuously monitored and recorded. |
(続き) A warning system should be in place to instantly indicate and warn operators of any failure in the air supply or reduction of air pressure differences (below set limits for those identified as critical). The warning signal should not be overridden without assessment and a procedure should be available to outline the steps to be taken when a warning signal is given. Where alarm delays are set, these should be assessed and justified within the CCS. Other air pressure differences should be monitored and recorded at regular intervals. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
PART A: GENERAL GUIDANCE SUPPLIMENTARY PROVISIONS TO PIC/S GMP GUIDE PART I CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Production Areas 3.11 Where processes are not closed and there is exposure of the product to the immediate room environment without a subsequent microbial inactivation process, (e.g. during additions of supplements, media, buffers, gasses, manipulations) appropriate environmental conditions should be applied. |
(続き) For aseptic manipulations parameters in line with Annex 1 (i.e. Grade A with Grade B background) should be applied. The environmental monitoring program should include testing and monitoring of non-viable contamination, viable contamination and air pressure differentials. The monitoring locations should be determined having regards to the QRM principles. The number of samples, volume, and frequency of monitoring, alert and action limits should be appropriate taking into account the QRM principles. |
4 Premises 4.4 For the manufacture of sterile products, there are four grades of cleanroom/zone. Grade B: For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Air pressure differences should be continuously monitored. 4.16 Indicators of air pressure differences should be fitted between cleanrooms and/or between isolators and their background. Set points and the criticality of air pressure differences should be considered within the CCS. Air pressure differences identified as critical should be continuously monitored and recorded. |
(続き) A warning system should be in place to instantly indicate and warn operators of any failure in the air supply or reduction of air pressure differences (below set limits for those identified as critical). The warning signal should not be overridden without assessment and a procedure should be available to outline the steps to be taken when a warning signal is given. Where alarm delays are set, these should be assessed and justified within the CCS. Other air pressure differences should be monitored and recorded at regular intervals. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
7. Air filtration, airflow direction and pressure differentials 7.8 The pressure control and monitoring devices used should be calibrated. Compliance with specifications should be regularly verified and the results recorded. |
7. Air filtration, airflow direction and pressure differentials (略) The pressure control and monitoring devices used should be calibrated and, where possible, be linked to an alarm system set according to the determined levels. |
9. Premises 9.9 The HVAC system and pressure cascade design for the different areas should be appropriately designed and maintained, in order to minimize the risk of product contamination and to protect personnel from the risks of radiation exposure. The pressure differentials should be controlled, monitored and recorded. Appropriate controls should be put in place to promote the containment of radioactivite gases and vapours. |
4. Premises 4.4 Four grades of cleanrooms or zones are normally used for the manufacture of sterile products. Grade B. For aseptic preparation and filling, this is the background cleanroom for grade A (where it is not an isolator). Where applicable, air pressure differential between grade B and an adjacent area should be continuously monitored. 4.16 Indicators of air pressure differential should be fitted between cleanrooms and between isolators and their background. Set points and the criticality of air pressure differential should be considered within the CCS. Air pressure differentials identified as critical should be continuously monitored and recorded. |
(続き) A warning system should be in place to instantly indicate and warn operators of any failure in the air supply or reduction of air pressure differential (below set limits for those identified as critical). The warning signal should not be overridden without appropriate assessment and a procedure should be available to outline the steps to be taken when a warning signal is given. Where alarm delays are set, these should be assessed and justified within the CCS. Other air pressure differentials should be monitored and recorded at regular intervals. |
Glossary contamination control strategy (CCS). A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・製造上の無菌性確保において特に重要と考えられる箇所の差圧については、常時監視行うこと、及び警報を要すること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・清浄度レベルの異なる区域間の差圧については監視すること。 ・滅菌前製品の管理上重要な差圧についての常時監視・警報。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードBにおける差圧の連続モニタリングおよび差圧計の設置箇所について。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 *【誤記訂正】厚生労働省通知の和訳ではここを ”CSS” と表記されていますが、PIC/Sの原文は ”CCS”であるため、誤記です。表は正しい ”CCS”を記載。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・重要な差圧、連続モニタリング・警告信号について。 ・CCSの評価によるアラーム遅延設定の妥当性について。 ・その他の差圧については定期的な監視・記録すること。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCSの定義。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・閉鎖系ではなく、微生物不活化処理を経ずに製品が直接室内環境に露出するプロセスの差圧の試験と環境モニタリングについて。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・クリーンルーム間の差圧の常時モニタリング。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・例として、施設の異常を検知するための差圧の仕様について。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4、4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.11と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.11と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4、4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・差圧の制御・監視装置について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・差圧の制御・監視装置について。 |
対象医薬品:放射線医薬品 以下の要件があります。 ・所定の室間差圧が制御・監視・記録されていること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4、4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | --- | --- | --- | --- | 欧州 | 欧州 | --- | --- | --- | --- | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | WHO | WHO | ||||||||||||||||||||||||||
| 分 類 | 省令 | 厚生労働省医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長 | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||||||||||||||
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 | 「PIC/S のGMP ガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3)PIC/S GMPガイドライン アネックス2A ヒト用先端医療医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3)PIC/S GMPガイドライン アネックス2B ヒト用生物学的医薬品( 原薬及び製剤) の製造 |
PIC/SのGMPガイドラインを活用する際の考え方について 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
PIC/SのGMPガイドラインを活用する際の考え方についての一部改正について 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性医薬品の製造 |
原薬GMPのガイドラインについて | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 2A MANUFACTURE OF ADVANCED THERAPY MEDICINAL PRODUCTS FOR HUMAN USE | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 2B MANUFACTURE OF BIOLOGICAL MEDICINAL SUBSTANCES AND PRODUCTS FOR HUMAN USE | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS Annex 3 MANUFACTURE OF RADIOPHARMACEUTICALS | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS Annex 7 MANUFACTURE OF HERBAL MEDICINAL PRODUCTS | EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 3 Manufacture of Radiopharmaceuticals |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 7 Manufacture of Herbal Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles1 1 The current document is a revision of WHO Good manufacturing practices for pharmaceutical products: main principles, previously published in WHO Technical Report Series, No. 961, 2011, Annex 3. |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 44th report Annex 3 WHO good manufacturing practices for pharmaceutical products containing hazardous substances WHO Technical Report Series, No. 957, 2010 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
||||||||||||||||||||||||||
| 発行年 | 2021年 | 2017年 | 2022年 | 2022年 | 2012年 | 2013年 | 2001年 | 2023年 | 2023年 | 2023年 | 2023年 | 2008年 | 2008年 | 2014年 | 2010年 | 2018年 | 2018年 | ||||||||||||||||||||||||||
| 記載内容 | 第二章 医薬品製造業者等の製造所における製造管理及び品質管理 第一節 通則 (交叉汚染の防止) 第八条の二 製造業者等は、医薬品に係る製品の交叉汚染を防止するため、製造手順等について所要の措置をとらなければならない。 (構造設備) 第九条 医薬品に係る製品の製造所の構造設備は、次に定めるところに適合するものでなければならない。 五 次に掲げる場合においては、製品等を取り扱う作業室(密閉容器に収められた製品等のみを取り扱う作業室及び製品等から採取された検体のみを取り扱う作業室を除く。次項において同じ。)を専用とし、かつ、空気処理システムを別系統にする等の当該製品等の漏出を防止する適切な措置がとられていること。 |
イ 飛散しやすく、微量で過敏症反応を示す製品等を取り扱う場合 ロ 交叉汚染することにより他の製品等に重大な影響が及ぶおそれのある製品等(強い薬理作用又は毒性を有するものを含む。)を取り扱う場合であって、交叉汚染を防止する適切な措置をとることができない場合 |
第5 章 製造 製造における交叉汚染の防止 5.19. 例えば以下のような適切な技術的又は組織上の手段によって、交叉汚染を防止すること。 (略) b) 適切なエアロック及び排気設備を備える。 c) 未処理若しくは処理が不十分な空気の循環又は再流入により引き起こされる汚染リスクを最小化する。 |
パートA : 一般的ガイダンス 第3 章 建物及び設備 建物 製造区域 3.1 製造設備の適切な設計及び運用により、全ての製品について交叉汚染を防止すること。製品品質に対するリスクに相応した交叉汚染防止措置を講じること。QRMの原則を用いて、当該リスクを評価及び管理すること。 ある種のATMPs及びその製造に係る原材料がもたらすリスク(例: ウイルス)のレベルに応じて、製造・包装作業に係る建物及び設備を専用化して、当該リスクを管理することが必要な場合がある。 |
3.2 QRMの原則の下で妥当性を示すことができる適切な運用上の措置及び/又は技術的措置を一連の製造ステップ全体にわたって適用することで、同一区域内で2 つ以上の異なるATMPs/バッチを同時に製造することが許容される場合があり得る。例えば: (b) 同じ室内で複数のアイソレータを使用して異なるウイルスベクターを加工するときには、その部屋及び当該設備から100% 排気されている(すなわち、再循環がない)こと。 |
3.4 隔離すべき感染性ウイルスベクター(例:腫瘍溶解性ウイルス、複製可能なベクター) を扱う製造活動の場合においては、文書化された汚染管理ストラジー(以下「CCS」)及びQRMの原則に基づいて、特別な注意事項を講じること。製造業者は、当該CCS及びQRMの原則に基づいて、必要とされる隔離レベルの妥当性を示すこと。建物及び設備を特定の製品に専用化する必要性及びその範囲は、QRMプロセスの結果により決定すること。場合によっては、国ごとの法律に従って、専用施設、専用区域又は専用設備が必要とされることがある。 | 3.5 異なる製造区域間の交叉汚染のリスクを最小化するよう、空気処理ユニットを設計し、構築し、かつ維持すること。また、ある区域に特化した空気処理ユニットが必要な場合がある。シングルパス空気システムの使用を、QRMの原則に基づいて、検討すること。 | 3.7 無菌製品を加工するには、陽圧管理区域を使用すること。ただし、封じ込めの理由から、病原体が露出する特定区域内の陰圧管理は許容される。特定のリスクのある原材料( 例: 病原体) の無菌処理に陰圧管理区域又はBSCを使用する場合には、適切な清浄グレードの陽圧管理ゾーンをその周囲に設けること。それらの気圧カスケードを明確に規定するとともに、アネックス1 に規定されているような適切なアラーム設定で継続的にモニターすること | 第5 章 製造 製造における交叉汚染の防止 5.12 根拠に基づくQRMプロセスを用いて、製造する製品の交叉汚染リスクを評価し、管理すること。考慮に入れる要素には、以下が含まれる:(略) QRMプロセスの結果に基づいて、当該プロセスのワークフロー及び特定の1 製品に建物及び設備を専用化する又は単回使用システムを用いる必要性及び範囲を確定させること。特定の製品に接触する部位を専用化する、又は製造施設全体を専用化することも含まれ得る。 5.18 特定されたリスクに対して適切な交叉汚染防止措置を整えておくこと。交叉汚染を防止するため検討し得る措置には、とりわけ以下が含まれる: (e) 潜在的な空中汚染物質を特定区域内に封じ込めるよう、エアロック及び気圧カスケードを用いる。 |
パートA . 一般的ガイダンス 建物及び設備 9 .仕上げ(二次) 作業 について専用化された設備を要するかは、上記の考慮に加えて、その生物学的医薬品に特有の必要性、及び同一施設で扱われる他の製品(生物学的製品でないものを含む) の特性等の考慮に依拠することとなる。その他、仕上げ作業についての管理措置には、添加する順序を定めること、混合する速度、時間及び温度を管理すること、光に当たるのを制限すること並びに漏出時における封じ込め及び清浄化の手順を定めることが含まれ得る。 11. 異なる製造区域間の交叉汚染のリスクを最小化するよう、空気処理ユニットを設計し、構築し、維持すること。また、ある区域に特化した空気処理ユニットが必要な場合がある。シングルパス空気システムの使用を、QRMの原則に基づいて、検討すること。 |
12. 無菌製品を加工するには、陽圧管理区域を使用すること。ただし、封じ込めの理由から、病原体が露出する特定区域内の陰圧管理は許容される。特にリスクのある原材料(例: 病原体) の無菌処理に陰圧管理区域又は安全キャビネットを使用する場合には、適切な清浄グレードの陽圧管理ゾーンを周囲に設けること。それらの気圧カスケードを明確に規定するとともに、適切なアラーム設定を行い継続的にモニターすること。 | 建物及び設備 製造 34. 同じ作業区域(ホットセル、LAFユニットなど)での異なる放射性製品を同時に製造することは、交叉汚染や混同のリスクを最小限にするため避けなければならない。 |
建物 製造区域 5. 植物薬及び植物薬調製品の検体採取、秤量、混合及び加工作業を行う際に、粉塵が生じる場合は、例えば集塵装置や専用施設を使用するなど、容易に清掃ができ、交叉汚染が防止できるような具体的な対策を講じること。 |
4 構造及び設備 4.2 ユーティリティ 4.21 必要な場合には、適切な換気・空気ろ過・排気システムを設置すること。これらのシステムは、汚染及び交叉汚染のおそれを最小にするように設計し、設置し、また、製造の段階に即した、空気圧、微生物(適切であれば)、塵埃、湿度及び温度の管理装置を備えること。原薬が環境に暴露される区域では、特に注意を払うこと。 4.22 空気を製造区域に再循環させる場合には、汚染及び交叉汚染のおそれを最小限にするように適切な対策を取ること。 |
PART A: GENERAL GUIDANCE SUPPLIMENTARY PROVISIONS TO PIC/S GMP GUIDE PART I CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Production Areas 3.1 Cross-contamination should be prevented for all products by appropriate design and operation of manufacturing facilities. The measures to prevent cross-contamination should be commensurate with the risks to product quality. QRM principles should be used to assess and control the risks. Depending on the level of risk presented by some ATMPs and the materials involved in their production (for example, viruses), it may be necessary to dedicate premises and equipment for manufacturing and/or packaging operations to control the risk. |
3.2 Concurrent production of two or more different ATMPs/batches in the same area might be permitted due to adequate operational and/or technical control where justified under QRM principles applied across the entire sequence of manufacturing steps. For example: (b) When more than one isolator is used to process different viral vectors within the same room there should be 100% air exhaustion from the room and the facility (i.e. no recirculation). |
3.4 Special precautions should be taken in the case of manufacturing activities involving infectious viral vectors (e.g. oncolytic viruses, replication competent vectors) that should be segregated based on a documented CCS and QRM principles. The manufacturer should justify the level of segregation required based on the CCS and through QRM principles. The outcome of the QRM process should determine the necessity for and extent to which the premises and equipment should be dedicated to a particular product. In some cases, dedicated facilities, dedicated areas or dedicated equipment may be required in accordance with the national law. | 3.5 Air handling units should be designed, constructed and maintained to minimise the risk of cross-contamination between different manufacturing areas and may need to be specific for an area. Consideration, based on QRM principles, should be given to the use of single pass air systems. | 3.7 Positive pressure areas should be used to process sterile products, but negative pressure in specific areas at the point of exposure of pathogens is acceptable for containment reasons. Where negative pressure areas or BSCs are used for aseptic processing of materials with particular risks (e.g. pathogens), they should be surrounded by a positive pressure clean zone of appropriate Grade. These pressure cascades should be clearly defined and continuously monitored with appropriate alarm settings as defined by Annex 1. | CHAPTER 5 PRODUCTION Prevention of Cross-contamination in Production 5.12 An evidence-based QRM process should be used to assess and control the cross-contamination risks presented by the products manufactured. (中略)The outcome of the QRM process should be the basis for determining the process workflow and necessity for and extent to which premises and equipment should be dedicated or single use systems should be used for a particular product. 5.18 Measures to prevent cross-contamination appropriate to the risks identified should be put in place. Measures that can be considered to prevent cross-contamination include, among others: (e) use of air locks and pressure cascade to confine potential airborne contaminant within a specified area, |
PART A: GENERAL GUIDANCE PREMISES AND EQUIPMENT 9. For finishing (secondary) operations12, the need for dedicated facilities will depend on consideration of the above together with additional considerations such as the specific needs of the biological medicinal product and on the characteristics of other products, including any non-biological products, in the same facility. Other control measures for finishing operations may include the need for specific addition sequences, mixing speeds, time and temperature controls, limits on exposure to light and containment and cleaning procedures in the event of spillages. |
11. Air handling units should be designed, constructed and maintained to minimise the risk of cross-contamination between different manufacturing areas and may need to be specific for an area. Consideration, based on QRM principles, should be given to the use of single pass air systems. | 12. Positive pressure areas should be used to process sterile products but negative pressure in specific areas at the point of exposure of pathogens is acceptable for containment reasons. Where negative pressure areas or safety cabinets are used for aseptic processing of materials with particular risks (e.g. pathogens), they should be surrounded by a positive pressure clean zone of appropriate grade. These pressure cascades should be clearly defined and continuously monitored with appropriate alarm settings. | PRODUCTION 34. Production of different radioactive products in the same working area (i.e. hot-cell, LAF unit), at the same time should be avoided in order to minimise the risk of cross-contamination or mix-up. |
Premises & Equipment Production area 5. Specific provisions should be made during sampling, weighing, mixing and processing operations of herbal substances and herbal preparations whenever dust is generated, to facilitate cleaning and to avoid cross-contamination, as for example, dust extraction, dedicated premises, etc. |
PRODUCTION 34. Production of different radioactive products in the same working area (i.e. hotcell, LAF unit), at the same time should be avoided in order to minimise the risk of cross-contamination or mix-up. |
Premises & Equipment Production area 5. Specific provisions should be made during sampling, weighing, mixing and processing operations of herbal substances and herbal preparations whenever dust is generated, to facilitate cleaning and to avoid cross-contamination, as for example, dust extraction, dedicated premises, etc. |
12. Premises Production areas 12.30 Production areas should be effectively ventilated, with air-control facilities (including filtration of air to a sufficient level to prevent contamination and cross-contamination, as well as control of temperature and, where necessary, humidity) appropriate to the products handled, to the operations undertaken and to the external environment. These areas should be regularly monitored during both production and non-production periods to ensure compliance with their design specifications. |
16. Good practices in production 16.12 Cross-contamination should be avoided by taking appropriate technical or organizational measures, for example: (略) (c) providing appropriately designed airlocks, pressure differentials, and air supply and extraction systems; |
10. Air-handling units 10.4 Risk management principles should be applied to address the potential of cross-contamination where energy wheels are used. |
1. Introduction (略) HVAC system design influences architectural building design and layout, for example, with regard to airlock positions, doorways and lobbies. These in turn have an effect on room pressure, pressure differentials, pressure cascades, contamination and cross-contamination control. Therefore, the design of the HVAC system should be considered at the initial design stage of a pharmaceutical manufacturing plant. |
5. Design of HVAC systems and components
HVAC systems should be appropriately designed and managed throughout their life cycle. Documentation such as schematic drawings should be maintained to reflect the current situation. 5.1 Risk management principles should be applied for HVAC systems. This includes, but is not limited to, appropriate design, operation and monitoring, control of the climatic conditions and the prevention of contamination and cross-contamination. 5.5 Air intake and exhaust air terminals should be positioned in a manner in relation to one another that assists in preventing cross-contamination. |
6. Full fresh air systems and recirculation systems
6.2 HEPA filters may be installed (in the supply air stream or return air stream) to remove contaminants and thus prevent cross-contamination. The HEPA filters in such an application should have an EN 1822 classification of at least H13 or equivalent. 6.3 HEPA filters may not be required to control cross-contamination where evidence that cross-contamination would not be possible has been obtained by other robust technical means, or where the air-handling system is serving a single product facility. |
7. Air filtration, airflow direction and pressure differentials 7.1 Where different products are manufactured at the same time, i.e. in different areas or rooms in a multiproduct manufacturing site, measures should be taken to ensure that dust cannot move from one room to another. Facility design and layout, appropriate levels of filtration, airflow direction and pressure differentials can assist in preventing cross-contamination. |
9. Dust, vapour and fume control The discharge location of dust, vapours and fumes should be carefully considered to prevent contamination and cross-contamination. 9.3 The dust extraction points should be positioned in such a way as to prevent dust and powders dropping down from the extract point causing contamination or cross-contamination. |
4. Premises 4.1 Premises design Both the architectural design of the building and that of the HVAC system should be carefully considered when attempting to achieve the general objectives of preventing contamination and cross-contamination and ensuring an appropriate environment for the production and control of pharmaceutical products. It is important to ensure that the required environmental conditions, cleanliness and containment are achieved and maintained. |
5. Design of HVAC systems and components The HVAC system should be appropriately designed, taking into consideration the design of the facility, with various rooms or areas for storage of materials and inprocess materials or products, processing, and movement of materials, products and personnel. The required cleanliness classification should be achieved, as well as other parameters, such as air filtration, airflow velocity, air volumes, pressure differentials, temperature, relative humidity, viable and non-viable particle counts and containment. Conditions and limits should be specified, based on need. Manufacturers should determine and define limits for these. | (続き) These should be realistic, appropriate and scientifically justifiable at rest, in operation and as built at the time of design. In determining these, relevant factors and risks should be considered, including but not limited to possible failures of AHUs, seasonal variations, properties and types of materials and products, numbers of personnel and risks of cross-contamination. |
6. Full fresh air systems and recirculation systems Manufacturers may select to have full fresh air systems (for an example, see Fig. A2.8) or recirculate treated air supplied to production areas (in a full fresh air system, no air is recirculated; in recirculation systems, a defined percentage of the air is recirculated). In both cases, the air supplied to the production areas should be appropriately treated, to ensure that the environmental conditions specified are met and that the risks for contamination and cross-contamination are controlled. | (続き)(略) In both scenarios, appropriate levels of filtration should be applied, to prevent contamination and cross-contamination. Manufacturers should ensure that when high-efficiency particulate air (HEPA) filters are used, these are appropriately installed, not damaged and thus suitable for the intended use (see tests described in Section 12). |
7. Air filtration, airflow direction and pressure differentials Effective ventilation and appropriate levels of filtration are recommended in basic GMP guidelines. Manufacturers should determine which classes of filters should be used in ensuring that contaminants from outside are not introduced into manufacturing areas and that where recirculation systems are used, filtration of recirculated air is carried out effectively, to ensure that there is no risk of cross-contamination. Where different products are manufactured in different rooms in the same facility at the same time, appropriate controls should be in place to ensure containment and prevention of contamination and cross-contamination. | (続き)(略) Fig. A2.9 is a schematic diagram of an example of an air-handling system serving rooms with horizontal directional flow, vertical directional flow and turbulent flow, for rooms 3, 4 and 5, respectively. In these rooms, the HEPA filters are indicated to have been placed terminally mounted in the rooms and not in the AHU. Whenever HEPA filters are terminally mounted, it can assist with preventing cross-contamination from room to room in the event of a fan failure. |
9. Dust, vapour and fume control (略) A dust extractor should normally not serve different rooms where different products can be processed at the same time, owing to the risks such as backflow or flow from room to room, resulting in possible contamination and cross-contamination. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・作業室の空気処理システムを別系統にする等。 |
対象医薬品:全般 以下の要件があります。 ・空気処理システムを別系統にする等の場合について。 |
対象医薬品:全般 以下の要件があります。 ・適切なエアロック及び排気設備といった手段、未処理若しくは処理が不十分な空気の循環又は再流入による汚染リスクについて。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・建物及び設備の専用化について。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・同一室内での複数のアイソレータの使用に関して、100%排気(再循環をしない方式※1)について。 ※1: 該当室から排出される空気を空調機に戻す(循環をおこなう)ことをしないでワンパスとすることを規定している。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・ 建物及び設備を特定の製品に専用化について。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・ 異なる製造区域間の交叉汚染リスクを最小化するような空気処理ユニット、ある区域に特化した空気処理ユニット、シングルパス空気システムについて。 *シングルパス空気システム:該当室から排出される空気を空調機に戻す(循環をおこなう)ことをしないでワンパスとすることを規定している。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・封じ込めにおける陽圧管理区域と陰圧管理、気圧カスケードについて。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 以下の要件があります。 ・ 建物及び設備の専用化、特定の製品に接触する部位を専用化、又は製造施設全体を専用化、エアロック及び気圧カスケードについて。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 以下の要件があります。 ・ 専用化された設備の必要性判断について、異なる製造区域間の交叉汚染のリスクを最小化する空気処理ユニット、ある区域に特化した空気処理ユニット、シングルパス空気システムについて。 *シングルパス空気システム:該当室から排出される空気を空調機に戻す(循環をおこなう)ことをしないでワンパスとすることを規定している。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 以下の要件があります。 ・封じ込めにおける陽圧管理区域と陰圧管理、気圧カスケードについて。 |
対象医薬品:放射性医薬品の製造 以下の要件があります。 ・ホットセル、ラミナーエアフロー(LAF)ユニットを用いた場合でも、異なる放射性製品を単一ユニット内で同時に製造することは、交叉汚染や混同のリスクから避けなければならない。 *ホットセル:放射性物質や放射線が漏れないように設計され、管理されている放射性物質を取り扱うための場所。 *LAFユニット:laminar air flow(層流)装置。クリーンベンチやクリーンブース等を示す。 |
対象医薬品:植物性医薬品 以下の要件があります。 ・粉じんが発生する工程の対策として集塵装置や専用施設など。 |
対象医薬品:原薬 以下の要件があります。 ・適切な換気・空気ろ過・排気システム、空気の再循環における対策について。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3章 建物及び設備 建物 製造区域 3.1と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3章 建物及び設備 建物 製造区域 3.2と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3章 建物及び設備 建物 製造区域 3.4と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3章 建物及び設備 建物 製造区域 3.5と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第3章 建物及び設備 建物 製造区域 3.7と同じ内容となります。 |
対象医薬品:ヒト用先端医療医薬品(再生医療等製品) 別紙(3) PIC/S GMP ガイドライン アネックス2A ヒト用先端医療医薬品の製造 パートA 一般的ガイダンス 第5章 製造 製造における交叉汚染の防止 5.12、5.18と同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤) の製造 パートA 一般的ガイダンス 建物及び設備 9と同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤) の製造 パートA 一般的ガイダンス 建物及び設備 11と同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品( 原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤) の製造 パートA 一般的ガイダンス 建物及び設備 12と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 建物及び設備 製造 34と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 製造区域 5.と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 建物及び設備 製造 34と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 製造区域 5.と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・製造区域において適切な空調設備を使用して効果的に換気すること。 ・監視する期間について。 |
対象医薬品:全般 以下の要件があります。 ・エアロック及び気圧カスケード、給気排気システムについて。 |
対象医薬品:有害物質含有医薬品 以下の要件があります。 ・energy wheelsの使用においては交叉汚染のリスクマネジメントを図ること。 *energy wheels:ロータリー型熱回収装置、回転式全熱交換器とも。回転式ローターにより排気に含まれる熱を回収して外気処理における熱負荷を削減する空調装置。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空気処理システムの設計差圧カスケードについて。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調システムにおけるリスクマネジメント適用、(外壁における)空気取り入れ口と排出口の配置について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・全外気空調方式と循環空調方式に関連して、HEPAフィルタについて(EN 1822規格におけるH13クラスを含む)。 *H13クラスについては、GMP構造設備要求比較 空気調和設備 .006:HEPAフィルタ設置要件を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・フィルタのレベル設定、気流方向と差圧について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・排気の屋外への放出口(排気ガラリや排気フード等)の配置や(室内における)集塵吸込み位置。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・建築設計における建物と空調設備において、交叉汚染を防止するよう考慮すること、清浄度や封じ込めの要求仕様の達成と維持について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調設備のシステムと装置類の設計における空調条件(空気ろ過や風速、風量、圧力差、温度、湿度、微生物および非微生物粒子数、封じ込め、清浄度クラス)と限度値について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 稼働時、非稼働時の空調条件や限度値の確定に関連する要因やリスクについて。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調システムにおける全外気方式と循環方式について、また適切なレベルのフィルタを設置することについて。 |
対象医薬品:非無菌医薬品 以下の要件があります。 汚染、交叉汚染防止ための適切なフィルターとして、HEPAフィルターの使用について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・フィルタの等級について、またHEPAフィルタを設置する場所(空調機内かダクト吹出口か)について。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・Fig.A2.9に,空気の流れ方向、HEPAフィルタの設置場所の例。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・異なる製品を製造している複数室に置ける集塵排気。 |
| 国 | 日本 | 日本 | 米国 | --- | 欧州 | --- | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | EUROPEAN COMMISSION | WHO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines for Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2011年 | 202 colspan="18"5年 | 2004年 | 2023年 | 2022年 | 2022年 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 記載内容 | 7.無菌医薬品に係る製品の作業所 7.2 空調システム (略) 空調システム及びその管理プログラムの基本要素には,温度,相対湿度,風量,換気回数,一方向気流,室間差圧,HEPAフィルターの完全性,浮遊微粒子数,微生物等が含まれる. 7.2.2 空気 (略) 4)重要区域(グレードA)の気流は,一方向気流とし,浮遊微粒子を区域外へ速やかに排出できるような流速及び均一性を有すること.また,近接する直接支援区域(グレードB)からの逆流のない気流を維持し,汚染を防止すること. 従来型の開放系クリーンブースやRABSを使用する場合,0.45m/sec±20%の平均風速が推奨される.アイソレータや特殊な適用事例においては,より遅い風速が適切な場合もある. |
(続き) 6) 一方向気流の定めがある場合においては,風速の変化が気流パターンに影響を及ぼす可能性があるので,プログラムに従って定められた間隔により各HEPA フィルターの吹出し風速についてモニタリングを行い,定められた風速が維持されていることを確認すること. |
2.用語の定義又は説明 2.2 アクセス制限バリアシステム(RABS: Restricted Access Barrier System):グローブを備えたハードウォールなどの物理的な障壁と,HEPAフィルターを介して供給される一方向気流,適切な管理運用システム等を主要な要素とするハードとソフトを融合した無菌操作区域(重要区域)を有するシステムをいう. 2.3 一方向気流(unidirectional airflow): ほぼ平行な流線で,一様な速度で流れるように調整された空気流をいう. |
18. 凍結乾燥工程 18.1 一般要件 2)凍結乾燥庫への搬入に際しては,重要区域(グレードA)の清浄度レベルが維持された作業環境で行うこと.トンネル型の自動搬送ライン,一方向気流装置を備えた搬送車,アイソレータ等,職員の直接的な介入を避ける方法を採用することが望ましい. |
4 建物 4.4 無菌製品の製造には、クリーンルーム/ 区画について4 つの清浄度等級がある。 グレードA:高リスク作業のための重要区画( 例: 無菌操作ライン、容器充填区画、止栓ボウル、開口状態の1次包装又はファーストエアの保護下で無菌接続を行う区画) 。通常、当該条件は、局所的な気流保護(RABS又はアイソレータ内での一方向気流作業台等)によって得られる。一方向気流が保たれていることが、グレードA 区域全体にわたって実証され且つ適格性評価されていること。 |
4.15 クリーンルーム及び清浄区画内での気流パターンを視覚化して、低い等級から高い等級の区域へ流入することがない旨、また、低い等級の区域( 床など) から又はより高い等級の区域へ汚染を運ぶおそれのある作業者又は設備の上を空気が漂うことのない旨を、実証すること。一方向気流を要する場合には、視覚化検討試験を行って適合を判定すること。( 4 . 4 節及び4 . 1 9 節を参照) | バリア技術 4.19 i . アイソレータ a . 開口式アイソレータの設計では、重要区画内においてファーストエアで保護され、操作の際に露出している製品の上を一方向気流が吹き流れるグレードA 条件を確保すること。 |
(続き) b. 閉鎖式アイソレータの設計では、操作の際に露出している製品を十分に保護するグレードA 条件を確保すること。単純な作業が実施される閉鎖式アイソレータ内においては、気流が完全に一方向でないことがあり得るが、露出した製品の汚染リスクを乱動気流で増大させてはならない。閉鎖式アイソレータ内に操作ラインを収容する場合には、重要区画内においてファーストエアで保護され、且つ操作の際に露出している製品の上を一方向気流が吹き流れるグレードA 条件を確保すること。 |
(続き) ii. RABS R A B S の設計は、重要区画内において 一方向気流及びファーストエアで保護されているグレードA 条件を確保するものであること。重要区画から周囲のバックグラウンド環境への陽圧気流が保たれていること。 |
4.20 i . アイソレータ b . アイソレータについてのC C S のリスク評価を行う際の主要な検討事項には、以下を含めること( ただし、これらに限定されるものではない): バイオ除染プログラム、自動化の程度、重要工程ポイントの「ファーストエア」保護を損なうおそれのある手袋操作のインパクト、バリア/手袋の完全性が失われた場合のインパクト、用いられる搬送メカニズム、及び始動準備又は保守管理等の作業のうちアイソレータの最終的なバイオ除染前にそのドアを開けることを要し得るもの。追加的な工程リスクが特定された場合には、バックグラウンドをより高い等級とすることを検討すること( なお、C C S において適切に妥当性が示れているときには、この限りでない) |
クリーンルーム及び清浄空気設備の適格性評価 4.23、無菌製品の製造用のクリーンルーム及び一方向気流ユニット( U D A F s ) 、R A B S 及びアイソレータ等の清浄空気設備は、当該環境の所要の特性に応じて適格性評価されていること。扱われている製品又は原材料の汚染のリスクが最小化されるためには、製造作業毎に適切な環境清浄度レベルが要求される。「非作業時」及び「作業時」の状態における適切な清浄度レベルが保たれていること。 |
4.30 一方向気流システムによって供給さ れる空気の速度は、空気速度測定の箇所を含めて適格性評価の実施計画書中において明確に妥当性を示しておくこと。空気速度を設定し、測定し、維持管理して、作業ポジション( 例: 高リスク作業が行われる箇所、製品及び/又は構成物が露出している箇所) において製品及び開口状態の構成物が適切な一方向の空気の動きで保護されるようになっていることを確保すること。一方向気流システムは、当該作業ポジションにおいて毎秒0.36〜0.54m の範囲内( ガイダンス値) の均一な空気速度を供するものであること( C C S において科学的に妥当性が示されているときには、この限りでない) | 4.32 クリーンルーム及び清浄空気設備の適格性再評価が、所定の手順に従って定期的に行われていること。その適格性再評価には最低限、以下を含めること: v . 空気速度試験 ( 注: グレードB 、C 及びD については、C C S の一環として文書化されたリスク評価に従って、空気速度試験が行われていること。ただし、一方向気流が供給される容器充填区画( 例:最終滅菌法による製品を容器充填する際、又はグレードA 及びRABSのバックグラウンド)には、空気速度試験が要求される。気流が一方向でない等級については、回復試験の計測で速度試験を代替すること。 |
5 設備 5.9 微粒子計数器( サンプリング管を含む)は、適格性評価されていること。チューブの直径及び曲がり半径については、その製造元の推奨仕様を検討すること。チューブの長さは一般的に1 m以内であること( なお、妥当性が示されているときには、この限りでない) 、且つ屈曲箇所の数が最小化されていること。サンプリング管の長さが短い携帯式微粒子計数器は、等級分け目的で用いること。等速サンプリングヘッドは、一方向気流システムにおいて用いること。微粒子計数器は、適切な方向に向けて重要箇所にできるだけ近く配置して、採取検体が全体を反映するものであることを確保すること。 |
7 人員 7.18 無菌区域における作業のうち製造工程に重要でないものは、無菌作業が進行しているときには特に、最小限にとどめること。人員の動きはゆっくりと、管理されて規則正しいものとし、過度の激しい活動による微粒子及び微生物の極端な拡散を防止すること。無菌作業を実行している作業者は常に無菌操作技術を厳守して、低品質の空気を重要区画内に流入させるおそれのある空気の流れの変化を防止すること。重要区画近辺での動きを制限し、一方向( ファーストエア) 気流の経路の妨げとならないようにすること。気流可視化検討試験の照査を、教育訓練プログラムの一環として、検討すること。 |
8 製造及び特有の技術 凍結乾燥 8.126 ii. 部分的に栓がされた容器の凍結乾燥機への搬送は、常時グレードA 条件下で行うとともに、直接の作業者介在が最小化されるように設計された方法で取り扱うこと。コンベアシステム又は可搬式システム等の技術( 例:清浄空気搬送台車、可搬式一方向気流作業台)を用いて、部分的に栓がされた容器の搬送用システムの清浄度が維持管理されていることを確保すること。代替として、バリデーションで裏付けられる場合には、グレードA で閉塞されてグレードB 区域内にある間は再び開けられないトレイを使い、部分的にストッパーが取り付けられたバイアルを保護し得る( 例:適切に閉塞された箱) 。 |
用語解説 ファーストエア - フィルタ処理された空気で、重要区画に到達する前の空気を汚染するおそれのある露出した製品及び製品接触表面に接触する前に遮られていないものをいう。 一方向気流 - 重要工程区域又は試験区域から微粒子を再現性よく排除するよう単一方向に、頑健で均一な仕方で、且つ十分な速度で流れる気流。 一方向気流(UDAF)ユニット - フィルタ処理された一方向気流が供給されるキャビネット(以前はラミナー気流ユニット又はL A F と呼ばれていたもの) |
用語解説 汚染制御ストラテジー(CCS) - 微生物、エンドトキシン/発熱性物質及び微粒子を制御する計画一式であり、現行の製品及び工程の理解から導き出され、工程性能及び製品品質を保証するもの。当該制御には、原薬、添加剤並びに薬剤の原材料及び構成物、施設及び設備の操作条件、工程内管理、最終製品の規格、並びに付随するモニタリング及び管理の頻度及び方法に関連するパラメータ及び特性が含まれ得る。 |
用語解説 アイソレータ - 内部に再現性のあるバイオ除染を行うことが可能な筐体で、グレードA条件に合致する内部作業区画を有し、その内部を毀損することなく外部環境(例:クリーンルームの周囲の空気及び人員)から持続的に隔離状態にするもの。アイソレータには主要な2つの種類がある。 i. 閉鎖式アイソレータシステムは、周囲環境への開放部を用いずに、補助的設備への無菌接続部を介して原材料を搬送することにより、アイソレータの内部が外から汚染されないようにする。閉鎖システムは作業の間、密封状態に保たれる。 |
用語解説 アクセス制限バリアシステム(RABS) - 所定の空気品質条件(無菌操作にはグレードA)に合致する閉鎖された(だが完全密封ではない)環境を提供するシステムで、堅牢な壁で囲われた筐体と一体化した手袋を用いて、周囲のクリーンルーム環境からその内部を分離するシステム。RABSの内表面は殺芽胞剤で消毒及び除染される。作業者は、手袋、半身スーツ、RTPその他の一体化された搬送ポートを使用して、RABS内部に操作を実行したり原材料を搬入したりする。その設計によっては、ドアが開かれることは稀であり、予め厳密に定められた条件下でのみ開けられる。 |
用語解説 迅速搬送システム/ポート(RTP) - RABS又はアイソレータ内への物品搬送用のシステムで、重要区画へのリスクを最小化するもの。一例として、アルファ/ ベータのポートがある迅速搬送容器が挙げられる。 |
用語解説 等速サンプリングヘッド - 可能な限り空気の乱れが生じないように設計されていて、ノズルがなかったとしてもノズル面積を通過したであろう微粒子と同量( すなわち、検体採取口に入ってくる空気の平均速度がその場での気流の平均速度とほぼ同じ(±20 パーセント) になる検体採取条件)の微粒子がノズルに入るようになっているサンプリングヘッド。 |
IV. Buildings and Facilities A. Critical Area - Class 100 (ISO 5) (略) HEPA-filtered air should be supplied in critical areas at a velocity sufficient to sweep particles away from the filling/closing area and maintain unidirectional airflow during operations. The velocity parameters established for each processing line should be justified and appropriate to maintan unidirectional airflow and air quality under dynamic conditions within the critical area.脚注5 脚注5 A velocity of 0.45 meters/second (90 feet per minute) has generally been established, with a range of plus or minus 20 percent around the setpoint. Higher velocities may be appropriate in operations generating high levels of particulates. |
(続き)(略) In situ air pattern analysis should be conducted at the critical area to demonstrate unidirectional airflow and sweeping action over and away from the product under dynamic conditions. The studies should be well documented with written conclusions, and include evaluation of the impact of aseptic manipulations (e.g., interventions) and equipment design. Videotape or other recording mechanisms have been found to be useful aides in assessing airflow initially as well as facilitating evaluation of subsequent equipment configuration changes. |
D. Air Filtration 2. High-Efficiency Particulate Air (HEPA) (略) HEPA filter leak testing alone is insufficient to monitor filter performance. It is important to conduct periodic monitoring of filter attributes such as uniformity of velocity across the filter (and relative to adjacent filters). Variations in velocity can cause turbulence that increases the possibility of contamination. Velocities of unidirectional air should be measured 6 inches from the filter face and at a defined distance proximal to the work surface for HEPA filters in the critical area. |
(続き) Velocity monitoring at suitable intervals can provide useful data on the critical area in which aseptic processing is performed. The measurements should correlate to the velocity range established at the time of in situ air pattern analysis studies. HEPA filters should be replaced when nonuniformity of air velocity across an area of the filter is detected or airflow patterns may be adversely affected. |
E. Design The effect of equipment design on the cleanroom environment should be addressed. Horizontal surfaces or ledges that accumulate particles should be avoided. Equipment should not obstruct airflow and, in critical areas, its design should not disturb unidirectional airflow. |
V. PERSONNEL TRAINING, QUALIFICATION, & MONITORING A. Personnel Some of the techniques aimed at maintaining sterility of sterile items and surfacesinclude: ・ Move slowly and deliberately Rapid movements can create unacceptable turbulence in a critical area. Such movements disrupt the unidirectional airflow, presenting a challenge beyond intended cleanroom design and control parameters. The principle of slow, careful movement should be followed throughout the cleanroom. |
(続き) ・ Keep the entire body out of the path of unidirectional airflow Unidirectional airflow design is used to protect sterile equipment surfaces, container-closures, and product. Disruption of the path of unidirectional flow air in the critical area can pose a risk to product sterility. ・ Approach a necessary manipulation in a manner that does not compromise sterility of the product To maintain sterility of nearby sterile materials, a proper aseptic manipulation should be approached from the side and not above the product (in vertical unidirectional flow operations). Also, operators should refrain from speaking when in direct proximity to the critical area. |
X. LABORATORY CONTROLS A. Environmental Monitoring 4. Monitoring Methods b. Active Air Monitoring (略) Manufacturers should be aware of a device's air monitoring capabilities, and the air sampler should be evaluated for its suitability for use in an aseptic environment based on collection efficiency, cleanability, ability to be sterilized, and disruption of unidirectional airflow |
APPENDIX 1: ASEPTIC PROCESSING ISOLATORS B. Design 1. Airflow (略) Turbulent flow can be acceptable within closed isolators, which are normally compact in size and do not house processing lines. Other aseptic processing isolators employ unidirectional airflow that sweeps over and away from exposed sterile materials, avoiding any turbulence or stagnant airflow in the area of exposed sterilized materials, product, and container closures. |
C. Transfer of Materials/Supplies Some transfer ports might have significant limitations, including marginal decontaminating capability (e.g., ultraviolet) or a design that has the potential to compromise isolation by allowing ingress of air from the surrounding room. In the latter case, localized HEPA-filtered unidirectional airflow cover in the area of such a port should be implemented. Isolators often include a mousehole or other exit port through which product is discharged, opening the isolator to the outside environment. Sufficient overpressure should be supplied and monitored on a continuous basis at this location to ensure that isolation is maintained. |
APPENDIX 3: PROCESSING PRIOR TO FILLING AND SEALING OPERATIONS
A. Aseptic processing from early manufacturing steps (略) Procedures (e.g., aseptic connection) that expose a product or product contact surfaces should be performed under unidirectional airflow in a Class 100 (ISO 5) environment. The environment of the room surrounding the Class 100 (ISO 5) environment should be Class 10,000 (ISO 7) or better. Microbiological and airborne particle monitoring should be performed during operations. Microbial surface monitoring should be performed at the end of operations, but prior to cleaning. Personnel monitoring should be performed in association with operations. |
GLOSSARY Unidirectional flow- An airflow moving in a single direction, in a robust and uniform manner, and at sufficient speed to reproducibly sweep particles away from the critical processing or testing area. Laminar flow- An airflow moving in a single direction and in parallel layers at constant velocity from the beginning to the end of a straight line vector. |
4 Premises 4.4 For the manufacture of sterile products there are four grades of cleanroom/zone. Grade A: The critical zone for high-risk operations (e.g. aseptic processing line, filling zone, stopper bowl, open primary packaging or for making aseptic connections under the protection of first air). Normally, such conditions are provided by a localised airflow protection, such as unidirectional airflow workstations within RABS or isolators. The maintenance of unidirectional airflow should be demonstrated and qualified across the whole of the grade A area. |
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is no ingress from lower grade to higher grade areas and that air does not travel from less clean areas (such as the floor) or over operators or equipment that may transfer contamination to the higher-grade areas.Where unidirectional airflow is required, visualisation studies should be performed to determine compliance, (see paragraphs 4.4 & 4.19). | BARRIER TECHNOLOGIES 4.19 i . Isolators: a. The design of open isolators should ensure grade A conditions with first air protection in the critical zone and unidirectional airflow that sweeps over and away from exposed products during processing. |
(続き) b. The design of closed isolators should ensure grade A conditions with adequate protection for exposed products during processing. Airflow may not be fully unidirectional in closed isolators where simple operations are conducted. However, any turbulent airflow should not increase risk of contamination of the exposed product. Where processing lines are included in closed isolators, grade A conditions should be ensured with first air protection in the critical zone and unidirectional airflow that sweeps over and away from exposed products during processing. |
(続き) ii. RABS The design of RABS should ensure grade A conditions with unidirectional airflow and first air protection in the critical zone. A positive airflow from the critical zone to the supporting background environment should be maintained. |
4.20 i . Isolators: b . Key considerations when performing the risk assessment for the CCS of an isolator should include (but are not limited to); the bio-decontamination programme, the extent of automation, the impact of glove manipulations that may potentially compromise ‘first air’ protection of critical process points, the impact of potential loss of barrier/glove integrity, transfer mechanisms used and activities such as set-up or maintenance that may require the doors to be opened prior to the final bio-decontamination of the isolator. Where additional process risks are identified, a higher grade of background should be considered unless appropriately justified in the CCS. |
CLEANROOM AND CLEAN AIR EQUIPMENT QUALIFICATION 4.23 Cleanrooms and clean air equipment such as unidirectional airflow units (UDAFs), RABS and isolators, used for the manufacture of sterile products, should be qualified according to the required characteristics of the environment. Each manufacturing operation requires an appropriate environmental cleanliness level in the operational state in order to minimize the risk of contamination of the product or materials being handled. Appropriate cleanliness levels in the “at rest” and “operational” states should be maintained. |
4.30 The speed of air supplied by unidirectional airflow systems should be clearly justified in the qualification protocol including the location for air speed measurement. Air speed should be designed, measured and maintained to ensure that appropriate unidirectional air movement provides protection of the product and open components at the working position (e.g. where high-risk operations occur and where product and/or components are exposed). Unidirectional airflow systems should provide a homogeneous air speed in a range of 0.36 - 0.54 m/s (guidance value) at the working position, unless otherwise scientifically justified in the CCS. Airflow visualization studies should correlate with the air speed measurement. | 4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include at a minimum the following: v. air velocity test (Note: For grade B, C and D the air velocity test should be performed according to a risk assessment documented as part of the CCS. However, it is required for filling zones supplied with unidirectional airflow (e.g. when filling terminally sterilised products or background to grade A and RABS). For grades with non-unidirectional airflow, a measurement of recovery testing should replace velocity testing). |
5 Equipment 5.9 Particle counters, including sampling tubing, should be qualified. The manufacturer’s recommended specifications should be considered for tube diameter and bend radii. Tube length should typically be no longer than 1m unless justified and the number of bends should be minimized. Portable particle counters with a short length of sample tubing should be used for classification purposes. Isokinetic sampling heads should be used in unidirectional airflow systems. They should be oriented appropriately and positioned as close as possible to the critical location to ensure that samples are representative. |
7 Personnel 7.18 Activities in clean areas that are not critical to the production processes should be kept to a minimum, especially when aseptic operations are in progress. Movement of personnel should be slow, controlled and methodical to avoid excessive shedding of particles and organisms due to over-vigorous activity. Operators performing aseptic operations should adhere to aseptic technique at all times to prevent changes in air currents that may introduce air of lower quality into the critical zone. Movement adjacent to the critical zone should be restricted and the obstruction of the path of the unidirectional (first air) airflow should be avoided. A review of airflow visualisation studies should be considered as part of the training programme. |
8 Production and Specific Technologies LYOPHILIZATION 8.126 ii. The transfer of partially closed containers to a lyophilizer should be undertaken under grade A conditions at all times and handled in a manner designed to minimize direct operator intervention. Technologies such as conveyor systems or portable transfer systems (e.g. clean air transfer carts, portable unidirectional airflow workstations) should be used to ensure that the cleanliness of the system used to transfer the partially closed containers is maintained. Alternatively, where supported by validation, trays closed in grade A and not reopened whilst in the grade B area may be used to protect partially stoppered vials (e.g. appropriately closed boxes). |
Glossary First Air - Refers to filtered air that has not been interrupted prior to contacting exposed product and product contact surfaces with the potential to add contamination to the air prior to reaching the critical zone. Unidirectional airflow - An airflow moving in a single direction, in a robust and uniform manner, and at sufficient speed, to reproducibly sweep particles away from the critical processing or testing area. Unidirectional Airflow (UDAF) unit - A cabinet supplied with filtered unidirectional airflow (previously referred to as a Laminar Airflow Unit or LAF). |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Glossary Isolator - An enclosure capable of being subject to reproducible interior bio-decontamination, with an internal work zone meeting grade A conditions that provides uncompromised, continuous isolation of its interior from the external environment (e.g. surrounding cleanroom air and personnel). There are two major types of isolators: i. Closed isolator systems exclude external contamination of the isolator’s interior by accomplishing material transfer via aseptic connection to auxiliary equipment, rather than use of openings to the surrounding environment. Closed systems remain sealed throughout operations. |
Glossary Restricted Access Barrier System (RABS) - System that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A), and using a rigid-wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, RTPs and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened, and only under strictly pre-defined conditions. |
Glossary Rapid Transfer System/Port (RTP) - A System used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Glossary Isokinetic sampling head - A sampling head designed to disturb the air as little as possible so that the same particles go into the nozzle as would have passed the area if the nozzle had not been there (i.e. the sampling condition in which the mean velocity of the air entering the sample probe inlet is nearly the same (± 20 percent) as the mean velocity of the airflow at that location). |
4 Premises 4.4 For the manufacture of sterile products, there are four grades of cleanroom/zone. Grade A: The critical zone for high-risk operations (e.g. aseptic processing line, filling zone, stopper bowl, open primary packaging or for making aseptic connections under the protection of first air). Normally, such conditions are provided by a localised airflow protection, such as unidirectional airflow workstations within RABS or isolators. The maintenance of unidirectional airflow should be demonstrated and qualified across the whole of the grade A area. |
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is no ingress from lower grade to higher grade areas and that air does not travel from less clean areas (such as the floor) or over operators or equipment that may transfer contamination to the higher grade areas. Where unidirectional airflow is required, visualisation studies should be performed to determine compliance, (see paragraphs 4.4 & 4.19). | Barrier Technologies 4.19 i . Isolators: a. The design of open isolators should ensure grade A conditions with first air protection in the critical zone and unidirectional airflow that sweeps over and away from exposed products during processing. |
(続き) b. The design of closed isolators should ensure grade A conditions with adequate protection for exposed products during processing. Airflow may not be fully unidirectional in closed isolators where simple operations are conducted. However, any turbulent airflow should not increase risk of contamination of the exposed product. Where processing lines are included in closed isolators, grade A conditions should be ensured with first air protection in the critical zone and unidirectional airflow that sweeps over and away from exposed products during processing |
(続き) ii. RABS The design of RABS should ensure grade A conditions with unidirectional airflow and first air protection in the critical zone. A positive airflow from the critical zone to the supporting background environment should be maintained. |
4.20 i . Isolators: b . Key considerations when performing the risk assessment for the CCS of an isolator should include (but are not limited to); the bio-decontamination programme, the extent of automation, the impact of glove manipulations that may potentially compromise ‘first air’ protection of critical process points, the impact of potential loss of barrier/glove integrity, transfer mechanisms used and activities such as set-up or maintenance that may require the doors to be opened prior to the final bio-decontamination of the isolator. Whre additional process risks are identified, a higher grade of background should be considered unless appropriately justified in the CCS. |
Cleanroom and clean air equipment qualification 4.23 Cleanrooms and clean air equipment such as unidirectional airflow units (UDAFs), RABS and isolators, used for the manufacture of sterile products, should be qualified according to the required characteristics of the environment. Each manufacturing operation requires an appropriate environmental cleanliness level in the operational state in order to minimize the risk of contamination of the product or materials being handled. Appropriate cleanliness levels in the “at rest” and “operational” states should be maintained. |
4.30 The speed of air supplied by unidirectional airflow systems should be clearly justified in the qualification protocol including the location for air speed measurement. Air speed should be designed, measured and maintained to ensure that appropriate unidirectional air movement provides protection of the product and open components at the working position (e.g. where high-risk operations occur and where product and/or components are exposed). Unidirectional airflow systems should provide a homogeneous air speed in a range of 0.36 - 0.54 m/s (guidance value) at the working position, unless otherwise scientifically justified in the CCS. Airflow visualization studies should correlate with the air speed measurement. | 4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include, at a minimum, the following: - Air velocity test (Note: For grade B, C and D the air velocity test should be performed according to a risk assessment documented as part of the CCS. However, it is required for filling zones supplied with unidirectional airflow (e.g. when filling terminally sterilised products or background to grade A and RABS). For grades with non-unidirectional airflow, a measurement of recovery testing should replace velocity testing).BA7 |
5 Equipment 5.9 Particle counters, including sampling tubing, should be qualified. The manufacturer’s recommended specifications should be considered for tube diameter and bend radii. Tube length should typically be no longer than 1m unless justified and the number of bends should be minimized. Portable particle counters with a short length of sample tubing should be used for classification purposes. Isokinetic sampling heads should be used in unidirectional airflow systems. They should be oriented appropriately and positioned as close as possible to the critical location to ensure that samples are representative. |
7 Personnel 7.18 Activities in clean areas that are not critical to the production processes should be kept to a minimum, especially when aseptic operations are in progress. Movement of personnel should be slow, controlled and methodical to avoid excessive shedding of particles and organisms due to over-vigorous activity. Operators performing aseptic operations should adhere to aseptic technique at all times to prevent changes in air currents that may introduce air of lower quality into the critical zone. Movement adjacent to the critical zone should be restricted and the obstruction of the path of the unidirectional (first air) airflow should be avoided. A review of airflow visualisation studies should be considered as part of the training programme. |
8 Production and Specific Technologies Lyophilization 8.126 ii. The transfer of partially closed containers to a lyophilizer should be undertaken under grade A conditions at all times and handled in a manner designed to minimize direct operator intervention. Technologies such as conveyor systems or portable transfer systems (e.g. clean air transfer carts, portable unidirectional airflow workstations) should be used to ensure that the cleanliness of the system used to transfer the partially closed containers is maintained. Alternatively, where supported by validation, trays closed in grade A and not reopened whilst in the grade B area may be used to protect partially stoppered vials (e.g. appropriately closed boxes). |
Glossary First Air - Refers to filtered air that has not been interrupted prior to contacting exposed product and product contact surfaces with the potential to add contamination to the air prior to reaching the critical zone. Unidirectional airflow - An airflow moving in a single direction, in a robust and uniform manner, and at sufficient speed, to reproducibly sweep particles away from the critical processing or testing area. Unidirectional Airflow (UDAF) unit - A cabinet supplied with filtered unidirectional airflow (previously referred to as a Laminar Airflow Unit or LAF). |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Glossary Isolator - An enclosure capable of being subject to reproducible interior bio-decontamination, with an internal work zone meeting grade A conditions that provides uncompromised, continuous isolation of its interior from the external environment (e.g. surrounding cleanroom air and personnel). There are two major types of isolators: i. Closed isolator systems exclude external contamination of the isolator’s interior by accomplishing material transfer via aseptic connection to auxiliary equipment, rather than use of openings to the surrounding environment. Closed systems remain sealed throughout operations. |
Glossary Restricted Access Barrier System (RABS) - System that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A), and using a rigid-wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, RTPs and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened, and only under strictly pre-defined conditions. |
Glossary Rapid Transfer System/Port (RTP) - A System used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Glossary Isokinetic sampling head - A sampling head designed to disturb the air as little as possible so that the same particles go into the nozzle as would have passed the area if the nozzle had not been there (i.e. the sampling condition in which the mean velocity of the air entering the sample probe inlet is nearly the same (± 20 percent) as the mean velocity of the airflow at that location). |
4 Premises 4.4 Four grades of cleanrooms or zones are normally used for the manufacture of sterile products. Grade A. This is the critical zone for high-risk operations (for example, aseptic processing line, filling zone, stopper bowl, open primary packaging, or for making aseptic connections under the protection of first air). Normally, such conditions are provided by a localized airflow protection, such as unidirectional airflow work stations within RABS or isolators. The maintenance of unidirectional airflow should be demonstrated and qualified across the whole of the grade A area. Direct intervention (for example, without the protection of barrier and glove port technology) into the grade A area by operators should be minimized by premises, equipment, process and procedural design. |
4.15 Airflow visualization studies should demonstrate airflow patterns within cleanrooms and zones proving that there is no ingress from lower-grade to higher-grade areas and that air does not flow from less clean areas (such as the floor) or over operators or equipment, thus transferring contaminants to the higher-grade areas.Where unidirectional airflow is required, visualization studies should be performed to demonstrate compliance (refer to paragraphs 4.4 and 4.19). | Barrier technologies 4.19 i . Isolators: a. The design of open isolators should ensure grade A conditions with first air protection in the critical zone and unidirectional airflow that sweeps over and away from exposed products during processing. |
(続き) b. The design of closed isolators should ensure grade A conditions with adequate protection for exposed products during processing. Airflow may not be fully unidirectional in closed isolators where simple operations are conducted. However, any turbulent airflow should not increase the risk of contamination of the exposed product. Where processing lines are included in closed isolators, grade A conditions should be ensured with first air protection in the critical zone and unidirectional airflow that sweeps over and away from exposed products during processing. |
(続き) a. The design of RABS should ensure grade A conditions with unidirectional airflow and first air protection in the critical zone. A positive airflow from the critical zone to the supporting background environment should be maintained. |
4.20 i . Isolators: b .Key considerations when performing the risk assessment for the CCS of an isolator should include the biodecontamination programme, the extent of automation, the impact of glove manipulations that may potentially compromise first air protection of critical process points, the impact of otential loss of barrier or glove integrity, transfer mechanisms used, and activities such as set-up or maintenance that may require the doors to be opened prior to the final biodecontamination of the isolator. Where additional process risks are identified, a higher grade of background should be considered unless appropriately justified in the CCS. |
Cleanroom and clean air equipment qualification 4.23 Cleanrooms and clean air equipment used for the manufacture of sterile products, such as unidirectional airflow units, RABS and isolators, should be qualified. Each manufacturing operation requires an appropriate environmental cleanliness level in the operational state in order to minimize the risk of contamination of the materials or product being handled. The appropriate cleanliness levels in the at rest and operational states should be maintained. |
4.30 The speed of air supplied by unidirectional airflow systems should be clearly justified in the qualification protocol, including the location for air speed measurement. Air speed should be designed, measured and maintained to ensure that appropriate unidirectional air movement provides protection of the product and open components at the working position (for example, where high-risk operations occur and where product or components are exposed). Unidirectional airflow systems should provide a homogeneous air speed in a range of 0.36-0.54 metres per second (m/s) (guidance value) at the working level, unless otherwise scientifically justified in the CCS. Airflow visualization studies should correlate with the air speed measurement. | 4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include at a minimum the following: v. air velocity test. Note: For grade B, C and D, the air velocity test should be performed according to a risk assessment documented as part of the CCS. It is however, required for filling zones supplied with unidirectional airflow (for example, when filling terminally sterilized products or background to grade A and RABS). For grades with non-unidirectional airflow, a recovery test should replace velocity testing. |
5. Equipment 5.9 Particle counters, including sampling tubing, should be qualified. The manufacturer’s recommended specifications should be considered for tube diameter and bend radii. Tube length should typically be no longer than 1 m unless justified, and the number of bends should be minimized. Portable particle counters with a short length of sample tubing should be used for classification purposes. Isokinetic sampling heads should be used in unidirectional airflow systems. They should be oriented appropriately and positioned as close as possible to the critical location to ensure that samples are representative. |
7. Personnel 7.18 Activities in clean areas that are not critical to the production processes should be kept to a minimum, especially when aseptic operations are in progress. The movement of personnel should be slow, controlled and methodical to avoid excessive shedding of particles and organisms due to overvigorous activity. Operators performing aseptic operations should adhere to aseptic technique at all times to prevent changes in air currents that may introduce air of lower quality into the critical zone. Movement adjacent to the critical zone should be restricted and obstruction of the path of the unidirectional (first air) airflow should be avoided. A review of airflow visualization studies should be considered as part of the training programme. |
8 Production and Specific Technologies Lyophilization 8.126 ii. The transfer of partially closed containers to a lyophilizer are undertaken under grade A conditions at all times and handled in a manner designed to minimize direct operator intervention. Technologies such as conveyor systems or portable transfer system s(for example, clean air transfer carts, portable unidirectional airflow workstations) should be used to ensure that the cleanliness of the system used to transfer the partially closed containers is maintained. Alternatively, where supported by validation, trays closed in a grade A area and not reopened whilst in the grade B area may be used to protect partially stoppered vials (such as appropriately closed boxes). |
Glossary first air. Filtered air that has not been interrupted prior to contacting exposed product and product contact surfaces with the potential to add contamination to the air prior to reaching the critical zone. unidirectional airflow. An airflow moving in a single direction in a robust and uniform manner and at sufficient speed to reproducibly sweep particles away from the critical processing or testing area. unidirectional airflow unit. A cabinet supplied with filtered unidirectional airflow (previously referred to as a laminar airflow unit). |
Glossary contamination control strategy (CCS). A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Glossary isolator. An enclosure capable of being subject to reproducible interior biodecontamination, with an internal work zone meeting grade A conditions that provide uncompromised continuous isolation of its interior from the external environment (for example, surrounding cleanroom air and personnel). There are two major types of isolators: ■ Closed isolator systems exclude external contamination of the isolator’s interior by accomplishing material transfer via aseptic connection to auxiliary equipment rather than use of openings to the surrounding environment. Closed systems remain sealed throughout operations. |
Glossary restricted access barrier system (RABS). A system that provides an enclosed, but not fully sealed, environment meeting defined air quality conditions (for aseptic processing grade A) and using a rigid wall enclosure and integrated gloves to separate its interior from the surrounding cleanroom environment. The inner surfaces of the RABS are disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, rapid transfer systems or ports, and other integrated transfer ports to perform manipulations or convey materials to the interior of the RABS. Depending on the design, doors are rarely opened and only under strictly predefined conditions. |
Glossary rapid transfer system or port. A system used for the transfer of items into RABS or isolators that minimizes the risk to the critical zone. An example would be a rapid transfer container with an alpha/beta port. |
Glossary isokinetic sampling head. A sampling head designed to disturb the air as little as possible so that the same particles go into the nozzle as would have passed the area if the nozzle had not been there (that is, the sampling condition in which the mean velocity of the air entering the sample probe inlet is nearly the same (± 20%) as the mean velocity of the airflow at that location). |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・空調システムの基本要素のひとつとして一方向気流が含まれること ・ グレードAにおける一方向気流および風速について。 *「一方向気流」の定義は「2.用語の定義又は説明」において以下に説明されています。 ・一方向気流(unidirectional airflow): ほぼ平行な流線で,一様な速度で流れるように調整された空気流をいう。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・一方向気流システムにおける吹出し風速のモニタリング。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・一方向気流の定義。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・凍結乾燥庫への充填容器搬入において、一方向気流装置を備えた搬送車等の使用。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・ファーストエアにより無菌接続を行う区画を保護する等、グレードAにおいては一方向気流が維持されることが実証され適格性評価されていること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・一方向気流を要する場合には、視覚化検討試験を行って適合を判定すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・ファーストエアにより無菌接続を行う区画を保護する等、グレードAにおいては一方向気流が維持されることが実証され適格性評価されていること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・閉鎖式アイソレータにおいて単純な作業を行う場合は一方向気流ではない場合があるが、その場合でも乱流による汚染があってはならない。 ・閉鎖式アイソレータに内に操作ラインを収容する場合はファーストエアにより保護され一方向気流をによりグレードAを確保すること。 *用語ついての補足:「閉鎖式アイソレータ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・RABSはファーストエアにより保護され一方向気流によりグレードAを確保すること。 *用語ついての補足:「RABS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アイソレータのCCSのリスク評価において重要工程ポイントの「ファーストエア」保護を損なうおそれのある手袋操作のインパクト等が検討事項とすること。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 クリーンルーム・一方向気流ユニット・RABS・アイソレータ等における当該環境の所要の特性に応じた適格性評価。 ・各製造作業毎に汚染リスクを最小化するための適切な清浄度を設定。 ・「非作業時」及び「作業時」状態における適切な清浄度レベルの維持。 *用語ついての補足:「一方向気流ユニット」「RABS」「アイソレータ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・一方向気流システムによって供給さ れる空気速度の妥当性について ・製品及び開口状態の構成物が適切な一方向の空気の動きで保護されるようになっていること。 ・一方向気流システムは毎秒0.36〜0.54m の範囲内( ガイダンス値) の均一な空気速度を供すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードB 、C 及びD の適格性再評価においては、空気速度試験についてリスク評価に従って実施する。 ・一方向気流が供給される容器充填区画には空気速度試験が必要。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微粒子係数におけるサンプリングチューブについて1 m以内であること、屈曲箇所の数が最小化されていること、サンプリング管の長さが短い携帯式微粒子計数器は、等級分け目的で用いること。 ・一方向気流システムにおいては等速サンプリングヘッドを用いること。 *用語ついての補足:「等速サンプリングヘッド」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌区域における作業において、一方向(ファーストエア)気流の妨げにならないよう作業者の動きを制限すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・部分的に栓がされた容器の凍結乾燥機への搬送における可搬式システム等の技術の例としての”可搬式一方向気流作業台”について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・ファーストエアの定義。 ・一方向気流の定義。 ・一方向気流(UDAF)ユニットの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCSの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アイソレータの定義。 ・ 閉鎖式アイソレータシステムの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アクセス制限バリアシステム(RABS)の定義。 *用語ついての補足:「RTP」は本表アネックス1の用語解説の「迅速搬送システム/ポート(RTP)」を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・迅速搬送システム/ポート(RTP)の定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・等速サンプリングヘッド |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・重要区域において給気HEPAフィルタにより供給される空気は、充填/閉塞エリアから微粒子を押し流すために十分で、作業下でも一方向流を維持しうる風速とすべき ・個々の工程作業に応じた速度パラメータとすること。 (脚注:風速0.45m/sec±20%、もしくはそれ以上) |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・動的条件下における一方向流と気流パターンの解析について。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・HEPA フィルターの通過する風速均一性に変化がある場合、乱気流を起こし、汚染を引き起こす可能性があること。 ・重要区域のHEPAフィルターにおける一方向性の気流速度は、フィルター表面から6 インチ離し、作業面近傍の定められた距離で測定されるべき。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・気流測定において、気流パターン解析による速度範囲との相関関係について。 ・フィルターの気流通過速度に不均一性がみられたとき、または気流パターンが悪影響を受けた可能性のあるときは、HEPA フィルターを交換すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・クリーンルーム環境に及ぼす装置設計において、微粒子が堆積するような水平面や出っ張りがなく、気流・一方向流を妨害しないように設計されること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌性維持において、作業者はゆっくりと慎重な動作により一方向流を乱さないこと。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌性維持において、作業者の体の一部分でも一方向流に入らないこと。 ・無菌操作を行う時に製品上部からではなく側面から近づくこと(垂直方向の一方向流の場合)。 ・重要操作区域直近では会話を行わないこと。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・試験室管理における環境モニタリングにおい用いるエアーサンプラーについて、一方向流を乱さないことなどの無菌環境下での適性を評価すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌操作に用いるアイソレーターについては一方向流を採用すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・アイソレーターにおける物の移送ポートに局所的なHEPA フィルター付の一方向流カバーを設置することについて。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・クラス100(ISO5)環境の一方向流下で無菌操作を実施すること。 ・同周囲環境はクラス10,000(ISO7)もしくはそれ以上とすること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・一方向気流の定義。 ・ラミナーフローの定義。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 i aと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 RABS 4.19 ii. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 アイソレータ 4.20 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.23と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.30と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.18と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 凍結乾燥 8.126と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、閉鎖式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 等速サンプリングヘッド 内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 i aと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 ii. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.23と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.30と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.30と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.18と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 凍結乾燥 8.126と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、閉鎖式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 等速サンプリングヘッド 内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.4と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.15と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 i aと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.19 ii. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 バリア技術 4.20 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.23と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.30と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.30と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.18と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 8 製造及び特有の技術 凍結乾燥 8.126と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アイソレータ、閉鎖式アイソレータシステムと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 アクセス制限バリアシステム(RABS)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 迅速搬送システム/ポート(RTP)と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 等速サンプリングヘッドと同じ 内容となります。 |
| 国 | 日本 | 日本 | 米国 | ― | 欧州 | ― | ― | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | ORA | PIC/S | EUROPEAN COMMISSION | WHO | WHO | |||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Guide to inspections | GMP | GMP | GMP | GMP | |||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
High Purity Water Systems Guide to inspections of high purity water systems | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 55th report Annex 2 WHO good manufacturing practices: water for pharmaceutical use WHO Technical Report Series., No. 1033, 2021 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||||
| 発行年 | 2011年 | 2025年 | 1993年 | 2023年 | 2022年 | 2021年 | 2022年 | |||||
| 記載内容 | 【参考情報】 A2 製薬用水 以下に製薬用水の製造管理及び品質管理に関する基本的な考え方を示す. A2.1 製薬用水設備の基本設計の留意点 10) 主配管がチーズ管(T字管)などで分枝し,その先にバルブなどの閉止機構があるようなデッドレグは,水が滞留しやすくなるので,主管の中心から枝管の先の閉止機構までの距離が枝管内径の6倍以内にすること.可能であれば3倍以内が好ましい. |
6 ユーティリティ 給水システム 6.7 微生物汚染を防ぎ、適切な品質の水の信頼できる供給源を確保するように、水処理設備及び配水システムが設計され、組み立てられ、据え付けられ、就役され、適格性評価され、モニターされ、且つ保守管理されていること。微粒子、微生物の汚染/増殖及びエンドトキシン/発熱性物質の存在のリスクを最小化するよう措置を講じること(例: 配管に傾斜を付けて完全に排水できるようにして、デッドレグをなくす) 。システム中にフィルタが含まれている場合には、そのモニタリング及び保守管理に特別な注意を払うこと。生産された水は、関連する薬局方の現行のモノグラフに適合すること。 |
用語解説 デッドレグ - 循環していない配管(流体が停滞しているおそれのある箇所)で配管内径の3倍以上の長さであるもの。 |
IX. PIPING One common problem with piping is that of ""dead-legs"". The proposed LVP Regulations defined dead-legs as not having an unused portion greater in length than six diameters of the unused pipe measured from the axis of the pipe in use. |
XI. PURIFIED WATER SYSTEMS (前略)Figure 11 and Figure12 illustrate another purified water system which had some problems. Unlike most of the other systems discussed, this is a one-way and not recirculating system. A heat exchanger is used to heat the water on a weekly basis and sanitize the system. Actually, the entire system is a ""dead-leg."". |
6 Utilities WATER SYSTEMS 6.7 Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination/proliferation and endotoxin/pyrogen (e.g. sloping of piping to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant Pharmacopeia. |
Glossary Dead leg - Length of non-circulating pipe (where fluid may remain static) that is greater than 3 internal pipe diameters. |
6 Utilities Water systems 6.7 Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination/proliferation and endotoxin/pyrogen (e.g. sloping of piping to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant Pharmacopeia. |
Glossary Dead leg - Length of non-circulating pipe (where fluid may remain static) that is greater than 3 internal pipe diameters. |
8. System sanitization and bioburden control 8.6 Other control techniques to be considered may include: (略) ■Ensuring hygienic design, including the use of zero dead leg diaphragm valves where possible, and minimizing dead legs elsewhere. Areas of possible dead legs should be measured and calculated. |
6. Utilities Water systems 6.7 (略)Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination and proliferation, and endotoxin/pyrogen (for example, by sloping pipes to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant pharmacopoeia. |
Glossary dead leg. Length of non-circulating pipe (where fluid may remain static) that is greater than three internal pipe diameters. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・主管の中心から枝管の先の閉止機構までの距離が枝管の内径の6倍以内。可能であれは3倍以内が好ましい。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微粒子、微生物汚染/増殖、エンドトキシン/発熱性物質の存在リスクを最小化するための対策がとられること。(デッドレグをなくすこと。) |
対象医薬品:無菌医薬品 以下の要件があります。 ・デッドレグの定義。 |
対象医薬品:全般 以下の要件があります。 ・LVPのCGMPの法案で、「デッドレグ」は主管の中心からの未使用部の長さが、枝管の内径の6倍以下。 |
対象医薬品:全般 以下の要件があります。 ・図11、12にて、精製水の配管系における「デッドレグ」の例示。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 11 用語解説 デッドレグと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 11 用語解説 デッドレグと同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・可能であればゼロデッドレグとなるダイヤフラムバルブの採用を含み、デッドレグを最小にすること。 ・デッドレグの可能性ある箇所は寸法を計測し計算すること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 11 用語解説 デッドレグと同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | ― | 欧州 | ― | ― | |
|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO | WHO | |
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | |
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 | 無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 55th report Annex 2 WHO good manufacturing practices: water for pharmaceutical use WHO Technical Report Series., No. 1033, 2021 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|
| 発行年 | 2021年 | 2011年 | 2025年 | 2023年 | 2022年 | 2021年 | 2022年 | |
| 記載内容 | 第三章 医薬部外品製造業者等の製造所における製造管理及び品質管理 第三節 無菌医薬部外品の製造管理及び品質管理 (無菌医薬部外品の製造所の構造設備) 第二十三条 五 無菌医薬品に係る製品の製造に必要な蒸留水等を供給する設備は、異物又は微生物による蒸留水等の汚染を防止するために必要な構造であること。 |
【参考情報】 A2 製薬用水 以下に製薬用水の製造管理及び品質管理に関する基本的な考え方を示す. A2.1 製薬用水設備の基本設計の留意点 A2.1.2 注射用水製造設備 横引き配管には排水時及び滅菌時の水の滞留を防止できるように可能な限り1/100以上の勾配を設けること。 |
6 ユーティリティ 給水システム 6.7 微生物汚染を防ぎ、適切な品質の水の信頼できる供給源を確保するように、水処理設備及び配水システムが設計され、組み立てられ、据え付けられ、就役され、適格性評価され、モニターされ、且つ保守管理されていること。微粒子、微生物の汚染/増殖及びエンドトキシン/発熱性物質の存在のリスクを最小化するよう措置を講じること(例: 配管に傾斜を付けて完全に排水できるようにして、デッドレグをなくす) 。システム中にフィルタが含まれている場合には、そのモニタリング及び保守管理に特別な注意を払うこと。生産された水は、関連する薬局方の現行のモノグラフに適合すること。 |
6 Utilities WATER SYSTEMS 6.7 Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination/proliferation and endotoxin/pyrogen (e.g. sloping of piping to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant Pharmacopeia. |
6 Utilities Water systems 6.7 Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination/proliferation and endotoxin/pyrogen (e.g. sloping of piping to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant Pharmacopeia. |
8. System sanitization and bioburden control 8.6 Other control techniques to be considered may include: (略) ■Installing pipework in a manner to allow for full drainage, if required. A guidance figure for the slope is not less than 1/100. |
7. Good practices for water systems For purified water and bulk water for injection systems (略) ■The system should be installed to promote drainability with a recommended minimum slope of 1/100. |
6. Utilities Water systems 6.7 (略)Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination and proliferation, an endotoxin/pyrogen (for example, by sloping pipes to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant pharmacopoeia. |
| 概 要 | 対象医薬品:無菌医薬部外品 以下の要件があります。 ・汚染を防止するために必要な構造であること。 *具体的な構造、構成の記載はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・注射用水の配管は、排水時及び滅菌時の水の滞留を防止できるように可能な限り1/100以上の勾配を設けること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微粒子、微生物汚染/増殖、エンドトキシン/発熱性物質の存在リスクを最小化するための対策がとられること。(配管勾配を設けて完全に排水できること。) |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.7と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・配管の施工は必要に応じてドレンが完全に抜けるようにすること。配管勾配のガイダンス値は1/100以上とすること。 |
対象医薬品:全般 以下の要件があります。 ・精製水、注射用水の配管の施工は排水性を確保するため配管勾配は1/100以上とすること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.7と同じ内容となります。 |
| 国 | 日本 | 日本 | ― | 欧州 | ― | ― | ||
|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO | WHO | ||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | ||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 55th report Annex 2 WHO good manufacturing practices: water for pharmaceutical use WHO Technical Report Series., No. 1033, 2021 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||
| 発行年 | 2011年 | 2025年 | 2023年 | 2022年 | 2021年 | 2022年 | ||
| 記載内容 | 【参考情報】 A2 製薬用水 以下に製薬用水の製造管理及び品質管理に関する基本的な考え方を示す. A2.1 製薬用水設備の基本設計の留意点 A2.1.2 注射用水製造設備 ②注射用水は、微生物及び有機物の定着を防ぐために80℃以上に加温し、十分な乱流となる流速で常時循環することが望ましい。 |
6 ユーティリティ 給水システム 6.9 配水システム内の配管を通る水の流れを乱流状態に保って、微生物の付着及びその後のバイオフィルム形成のリスクを最小化すること。流量は、適格性評価の際に確立され、且つ日常的にモニターされていること。 |
6 Utilities WATER SYSTEMS 6.9 Water flow should remain turbulent through the pipes in water distribution systems to minimize the risk of microbial adhesion, and subsequent biofilm formation. The flow rate should be established during qualification and be routinely monitored. |
6 Utilities Water systems 6.9 Water flow should remain turbulent through the pipes in water distribution systems to minimize the risk of microbial adhesion, and subsequent biofilm formation. The flow rate should be established during qualification and be routinely monitored. |
7. Good practices for water systems 7.2.1 As a minimum, the following design and construction practices should be considered. For purified water and bulk water for injection systems ■ Provision should be made for in-line measurement for total organic carbon (TOC), conductivity, pressure, flow and temperature. |
8. System sanitization and bioburden control 8.6 Other control techniques to be considered may include: (略) ■The maintenance of a continuous circulation of water maintaining turbulent flow evidenced by, for example, a Reynolds number of > 4000. |
12. Continuous system monitoring 12.3 A combination of online and offline instruments, linked to appropriately qualified alarm systems, should be used. Parameters such as flow, pressure, and temperature should be monitored with online instruments - as well as conductivity and TOC, where possible. |
6. Utilities Water systems 6.9 Water flow should remain turbulent through the pipes in water distribution systems to minimize the risk of microbial adhesion and subsequent biofilm formation. The flow rate should be verified during qualification and be routinely monitored. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・配管内の注射用水は、十分乱流となる流速で常時循環することが望ましい。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微生物が付着し、バイオフィルムが形成されるリスクを最小化するために、配水管内の水流は乱流を保つこと。 ・流量は、適格性評価の際に確立され、且つ日常的にモニターされていること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.9と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・流量をインラインで測定できるようにすること。 |
対象医薬品:全般 以下の要件があります。 ・乱流となるレイノルズ数(Re)を4000以上に維持するように水の循環を行うこと。 |
対象医薬品:全般 以下の要件があります。 ・流量はオンラインで測定し、モニタリングすること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.9と同じ内容となります。 |
| 国 | 日本 | 日本 | 米国 | ― | 欧州 | ― | ― | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | ORA | PIC/S | EUROPEAN COMMISSION | WHO | WHO | ||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Guide to inspections | GMP | GMP | GMP | GMP | ||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
High Purity Water Systems Guide to inspections of high purity water systems | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 55th report Annex 2 WHO good manufacturing practices: water for pharmaceutical use WHO Technical Report Series., No. 1033, 2021 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||
| 発行年 | 2011年 | 2025年 | 1993年 | 2023年 | 2022年 | 2021年 | 2022年 | ||||||||||
| 記載内容 | 【参考情報】 A2 製薬用水 以下に製薬用水の製造管理及び品質管理に関する基本的な考え方を示す. A2.1.2 注射用水製造設備 ②注射用水は、微生物及び有機物の定着を防ぐために80℃以上に加温し、十分な乱流となる流速で常時循環することが望ましい。 |
6 ユーティリティ 給水システム 6.10 注射用水( W F I ) は、適格性評価の際に定められている規格に合致する水から生産し、微生物生育のリスクを最小化する方法( 例:70 ℃ を超える温度で常時循環させる) で貯蔵及び分配すること。W F I は、蒸留又は蒸留と同等の精製工程で生産すること。これには、電気脱脱イオン、限外濾過又はナノフィルタ処理等の他の適切な技術と組み合わせた逆浸透法が含まれ得る。 |
IX. PIPING (略)It should be pointed out that this was developed for hot 75 - 80°circulating systems. With colder systems (65 - 75℃), any drops or unused portion of any length of piping has the potential for the formation of a biofilm and should be eliminated if possible or have special sanitizing procedures. |
VII. HOLDING TANK In hot systems, temperature is usually maintained by applying heat to a jacketed holding tank or by placing a heat exchanger in the line prior to an insulated holding tank. |
X. REVERSE OSMOSIS As an additional comment on RO systems, with the recognition of microbiological problems, some manufacturers have installed heat exchangers immediately after the RO filters to heat the water to 75 - 80℃ to minimize microbiological contamination. |
XI. PURIFIED WATER SYSTEMS (略)It should be pointed out that simply because this is a one-way system, it is not inadequate. With good Standard Operational Procedures, based on validation data, and routine hot flushings of this system, it could be acceptable. A very long system (over 200 yards) with over 50 outlets was found acceptable. This system employed a daily flushing of all outlets with 80℃ water. |
6 Utilities WATER SYSTEMS 6.10 Water for injections (WFI) should be produced from water meeting specifications that have been defined during the qualification process, stored and distributed in a manner which minimizes the risk of microbial growth (e.g. by constant circulation at a temperature above 70°C). WFI should be produced by distillation or by a purification process that is equivalent to distillation. This may include reverse osmosis coupled with other appropriate techniques such as electrodeionization (EDI), ultrafiltration or nanofiltration. |
6 Utilities Water systems 6.10 Water for injections (WFI) should be produced from water meeting specifications that have been defined during the qualification process, stored and distributed in a manner which minimizes the risk of microbial growth (e.g. by constant circulation at a temperature above 70°C). WFI should be produced by distillation or by a purification process that is equivalent to distillation. This may include reverse osmosis coupled with other appropriate techniques such as electrodeionization (EDI), ultrafiltration or nanofiltration. |
4. Water quality specifications 4.3 Bulk purified water 4.3.4 The following are examples of aspects that should be considered when configuring a water purification system or defining URS: (略) ■unit process steps provided and documented with the appropriate instrumentation to measure parameters such as flow, pressure, temperature, conductivity and total organic carbon; 4.3.5 Ambient-temperature systems such as ion exchange and ultrafiltration are especially susceptible to microbiological contamination, particularly when equipment is static during periods of no or low demand for water. |
4.3.7 The following controls, for example, should be considered in order to minimize microbial contamination: ■control of temperature in the system, for example, by heat exchangers or room cooling in order to reduce the risk of microbial ghrowth; ;■the use of water-treatment system components that can periodically be thermally sanitized above 70℃ for a defined period of time, or chemically sanitized using, for example, ozone, hydrogen peroxide and/or peracetic acid; and ■ a combination of thermal and chemical sanitization, if required. |
5. General considerations for water purification systemsd 5.7 The specifications for water purification equipment, storage and distribution systems should take into account at least the following:d ■ the extremes in temperature that the system will encounter. |
7. Good practices for water systems 7.2.1 As a minimum, the following design and construction practices should be considered. For purified water and bulk water for injection systems ■ Provision should be made for in-line measurement for total organic carbon (TOC), conductivity, pressure, flow and temperature. |
8. System sanitization and bioburden control 8.2 Controls may include using chemical and/or thermal sanitization procedures as appropriate for production, storage and distribution systems. The procedure and conditions used (such as times and temperatures, as well as the frequency), should be defined and proven to be effective for sanitizing all relevant parts of the system. |
8.3 Systems that operate and are maintained at elevated temperatures (e.g. > 70 ℃) are generally less susceptible to microbiological contamination than systems that are maintained at lower temperatures. When lower temperatures are required due to the water treatment processes employed, or the temperature requirements for the water in use, special precautions should be taken to prevent the ingress of contaminants including microorganisms (see section 9.2 for guidance). 8.6 Other control techniques to be considered may include: ■Maintaining the system at an elevated temperature (e.g. > 70℃), if required. |
10. Water distribution 10.5 When the temperature is reduced for processing purposes, the reduction should occur for the minimum necessary time. The cooling cycles and their duration should be proven satisfactory during the qualification of the system. |
12. Continuous system monitoring 12.3 A combination of online and offline instruments, linked to appropriately qualified alarm systems, should be used. Parameters such as flow, pressure, and temperature should be monitored with online instruments - as well as conductivity and TOC, where possible. |
6 Utilities Water systems 6.10 Water for injection (WFI)) should be produced from water meeting specifications that have been defined during the qualification process, stored and distributed in a manner that minimizes the risk of microbial growth (for example, by constant circulation at a temperature above 70 °C). WFI should be produced by distillation or other suitable means. These may include reverse osmosis coupled with other appropriate techniques such as electrodeionization (EDI), ultrafiltration or nanofiltration. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・注射用水は、微生物及び有機物の定着を防ぐために80℃以上に加温し常時循環することが望ましい。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・微生物増殖のリスクを最小化する方法(例えば、70℃超える温度での連続循環)で保管・分配されること。 |
対象医薬品:全般 以下の要件があります。 ・高純水について、75〜80℃の高温循環を推奨している。また、65〜75℃の場合は、水滴や未使用箇所がバイオフィルムを形成する可能性があるため、可能であれば取り除く、または特別なサニタイゼーション手順があること。 |
対象医薬品:全般 以下の要件があります。 ・高温水系において、ジャケット付タンクに熱を供給するか、あるいは熱交換器を設置して保温材付きの貯水タンクに供給することにより温度維持を行うこと。 |
対象医薬品:全般 以下の要件があります。 ・逆浸透膜ROシステムにおいて、ROフィルタの後段直近に熱交換器を設け、微生物汚染を抑制するため75〜80℃まで昇温することがある。 |
対象医薬品:全般 以下の要件があります。 ・精製水システムにおいて、一方向流システムであるだけではなく、バリデーションデータに基づいたSOPに従った温水フラッシングが必要である。180m以上の配管長で50以上の供給末端がある場合などもあるが、日常的に80℃温水でのフラッシングを全ての末端において行うことになる。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.10と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・の製造設備において、温度等のパラメータの計測機器の設置を考慮すること。 ・精製水の製造におけるイオン交換,逆浸透法,限外ろ過等のシステムは環境温度下で用いられるため、停止時などは特に微生物汚染が発生しやすい。 |
対象医薬品:全般 以下の要件があります。 ・微生物増殖リスクを抑制するためには配管内温度を制御する熱交換器を設置するか、製造室の温度を制御すること。 ・水処理システムは定期的に、熱によるサニタイゼーションとして、70℃を超える温度で定められた時間を保持するか、オゾン等による化学的消毒を行うこと。必要に応じて温度と化学的消毒を併用すること。 |
対象医薬品:全般 以下の要件があります。 ・精製水システムにおける装置、貯蔵、配水ラインにおいておこりうる温度範囲に対する考慮をすること。 |
対象医薬品:全般 以下の要件があります。 ・温度をインラインで測定できるようにすること。 |
対象医薬品:全般 以下の要件があります。 ・システムの消毒及びバイオバーデン管理において、化学的および/または温度によるサニタイゼーションを含むこと。 手順と条件(時間、温度、頻度など)を定義し、システムの関連部分すべてをサニタイゼーションをするのに効果的であることを証明すること。 |
対象医薬品:全般 以下の要件があります。 ・高温(例:℃℃超過)で運転・維持されるシステムは、低温で維持されるシステムよりも、一般的に微生物汚染の影響を受けにくい。水処理プロセスや使用水の温度要件により製造用水の温度を低くしなくてはならない場合は、微生物を含む汚染を防ぐための措置を講じること。 ・必要に応じてシステムを高温(70℃以上)に保つこと。 |
対象医薬品:全般 以下の要件があります。 ・処理目的で用水供給温度を下げる場合、その時間は必要最小限に抑えること。冷却サイクルとその期間について、システムの適格性確認時に適切であることが証明されること。 |
対象医薬品:全般 以下の要件があります。 ・温度はオンラインで計測されること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.10と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | ― | 欧州 | ― | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO | ||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 薬局方 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | ||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 第十八改正日本薬局方 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||
| 発行年 | 2011年 | 2012年 | 2021年 | 2025年 | 2023年 | 2022年 | 2022年 | ||||||||||
| 記載内容 | 2.用語の定義又は説明 2.37 ピュアスチーム(pure steam): 精製水又はそれ以上の品質の水を用いて発生させた飽和蒸気であって、その凝縮水が注射用水の規格に適合するものをいう。 6. 最終滅菌法による医薬品の製造区域 6.3.2 高圧蒸気滅菌 1) 滅菌に用いる蒸気の品質は、滅菌対象物の本来の性能及び安全性を損なわないものであること。一般的には精製水又はそれ以上の品質の水を用いて発生させた蒸気(ピュアスチーム)を使用する。蒸気の凝縮水は、製品の仕込み水と同等以上の規格に適合すること。 定期的にその性状を確認し、品質の劣化が疑われる場合においては、原因を調査し適切な措置を採ること。 |
13.滅菌工程 13.2 高圧蒸気滅菌 1) 滅菌に用いる蒸気の品質は、滅菌対象物の本来の性能及び安全性を損なわないものであること。一般的には精製水又はそれ以上の品質の水を用いて発生させた蒸気(ピュアスチーム)を使用する。蒸気の凝縮水は、製品の仕込み水と同等以上の規格に適合すること。 定期的にその性状を確認し、品質の劣化が疑われる場合においては、原因を調査し適切な措置を採ること。 |
15. 無菌製造設備の定置蒸気滅菌(SIP) 15.1 2)SIPには、精製水又はそれ以上の品質の水を用いて発生させた蒸気を用いること。蒸気の凝縮水は、製品の仕込み水と同等以上の規格に適合すること。 |
11. 湿熱滅菌工程 高圧蒸気滅菌を含む湿熱滅菌は、無菌医薬品の最終滅菌手段として、最も広く採用されている滅菌法である。本章に述べる要件は、湿熱滅菌による滅菌を前提としたものであるが、基本的な考え方や手法の多くは、他の滅菌法にも共通に適用できるものである。 11.1 滅菌工程の設計 11.1.1 滅菌剤の特性 滅菌剤は、飽和蒸気又は湿熱(空気混合蒸気や過熱熱水)であり製品品質に悪影響を与える物質を含んでいてはならない。いずれの滅菌剤を使用する場合も微生物殺滅効果の有効性を確立し文書化すること。 |
参考情報 G4.微生物関連 滅菌法及び滅菌指標体〈G4-10-162〉 2. 滅菌法 2.1. 加熱法 2.1.1. 湿熱滅菌法 表1 湿熱滅菌における管理項目,ユーティリティ及び制御装置(参考) 管理項目/飽和蒸気滅菌 ・蒸気品質(過熱度,乾燥度,非凝縮性ガス濃度,必要に応じて化学的純度) |
6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.16 純水蒸気(清浄水蒸気) 発生装置への供給水は、適切に精製されたものであること。純水蒸気発生装置は、生成される水蒸気の品質が所定の化学物質及びエンドトキシンのレベルに合致していることを確保するように設計され、適格性評価され、運用されていること。 |
6.17 直接滅菌剤として用いられる水蒸気は、適切な品質のものであること、また、製品又は設備の汚染を引き起こし得るレベルの添加物を含有してはならない。原材料又は製品接触面( 例: 多孔質/硬質性のオートクレーブ載荷物) の直接滅菌用の純水蒸気を供給する発生装置については、水蒸気の凝縮液が関連する薬局方のW F I の現行モノグラフ( 微生物試験は水蒸気凝縮液に必須でない) に適合すること。 | (続き) 適切な検体採取スケジュールが整っていて、装置全体を反映する純水蒸気が定期的に分析に供されることを確保すること。滅菌用の純水蒸気の品質についてのその他事項として、バリデートされたパラメータに対して定期的な評価がなされていること。それらパラメータには以下を含めること( なお、別途妥当性が示されているときには、この限りでない) : 非凝縮性ガス、乾燥度(乾燥率)及び加熱度。 |
6 Utilities STEAM USED AS A DIRECT STERILISING AGENT 6.16 Feed water to a pure steam (clean steam) generator should be appropriately purified. Pure steam generators should be designed, qualified and operated in a manner to ensure that the quality of steam produced meets defined chemical and endotoxin levels. |
6.17 Steam used as a direct sterilising agent should be of suitable quality and should not contain additives at a level that could cause contamination of product or equipment. For a generator supplying pure steam used for the direct sterilisation of materials or product-contact surfaces (e.g. porous hard-good autoclave loads), steam condensate should meet the current monograph for WFI of the relevant Pharmacopeia (microbial testing is not mandatory for steam condensate). | (続き) A suitable sampling schedule should be in place to ensure that representative pure steam is obtained for analysis on a regular basis. Other aspects of the quality of pure steam used for sterilisation should be assessed periodically against validated parameters. These parameters should include the following (unless otherwise justified): non-condensable gases, dryness value (dryness fraction) and superheat. |
6 Utilities Steam used as a direct sterilising agent 6.16 Feed water to a pure steam (clean steam) generator should be appropriately purified. Pure steam generators should be designed, qualified and operated in a manner to ensure that the quality of steam produced meets defined chemical and endotoxin levels. |
6.17 Steam used as a direct sterilising agent should be of suitable quality and should not contain additives at a level that could cause contamination of product or equipment. For a generator supplying pure steam used for the direct sterilisation of materials or product-contact surfaces (e.g. porous hard-good autoclave loads), steam condensate should meet the current monograph for WFI of the relevant Pharmacopeia (microbial testing is not mandatory for steam condensate). | (続き) A suitable sampling schedule should be in place to ensure that representative pure steam is obtained for analysis on a regular basis. Other aspects of the quality of pure steam used for sterilisation should be assessed periodically against validated parameters. These parameters should include the following (unless otherwise justified): non-condensable gases, dryness value (dryness fraction) and superheat. |
6 Utilities 6.16 Steam used as a direct sterilizing agent Feed water to a pure steam (clean steam) generator should be appropriately purified. Pure steam generators should be designed, qualified and operated in a manner that ensures that the quality of steam produced meets defined chemical and endotoxin levels. |
6,17 Steam used as a direct sterilizing agent should be of suitable quality and should not contain additives at a level that could cause contamination of product or equipment. For a generator supplying pure steam used for the direct sterilization of materials or product contact surfaces (such as porous hard-good autoclave loads), steam condensate should meet the current monograph for WFI of the relevant pharmacopoeia (microbial testing is not mandatory for steam condensate). | (続き) A suitable sampling schedule should be in place to ensure that the sample for analysis is collected on a regular basis. The sample should be representative of the pure steam. Other aspects of the quality of pure steam used for sterilization should be assessed periodically against parameters. These parameters should include the following (unless otherwise justified): non-condensable gases, dryness value (dryness fraction) and superheat. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・ピュアスチームの定義。 ・高圧蒸気滅菌機の定義。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・高圧蒸気滅菌:飽和蒸気の原水は精製水かそれ以上の品質であること。 ・飽和蒸気の凝縮水の性状を定期的に確認し、品質の劣化が疑われる場合においては、原因を調査し適切な措置を採ること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・飽和蒸気の凝縮水は無菌医薬品の仕込み水と同等以上の規格に適合すること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・滅菌剤の特性:滅菌剤は、飽和蒸気又は湿熱(空気混合蒸気や過熱熱水)であり製品品質に悪影響を与える物質を含んでいてはならない。いずれの滅菌剤を使用する場合も微生物殺滅効果の有効性を確立し文書化すること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・湿熱滅菌法のうち、飽和蒸気滅菌における管理項目は、蒸気滅菌品質として過熱度、乾燥度、非凝縮性ガス濃度、必要に応じて化学的純度。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・生成される蒸気の品質が定義された化学物質およびエンドトキシンレベルを満たすことを保証する方法で設計され、評価され、運転されること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・製品又は設備の汚染を引き起こし得るレベルの添加物を含有してはならない。 ・蒸気凝縮物は関連薬局方のWFIに関する現行モノグラフを満たすべきである(蒸気凝縮物に対する微生物試験は必須ではない)。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・検体採取について。 ・純粋蒸気の品質のパラメータには、(特に正当な理由がない限り)非凝縮性ガス、乾燥度、過熱度が含まれること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.16と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.17と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.17と同じ内容となります。 |
| 国 | 日本 | 日本 | ― | 欧州 | ― | ||
|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO | ||
| 分 類 | 薬局方 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | ||
| 法規類 タイトル |
第十八改正日本薬局方 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products Premises |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||
| 発行年 | 2021年 | 2025年 | 2023年 | 2022年 | 2022年 | ||
| 記載内容 | 医薬品各条 注射用水 Water for Injection 本品は,「常水」にイオン交換,逆浸透等による適切な前処理を行った水又は「精製水」の,蒸留又は超ろ過により製したものである。本品を超ろ過法(逆浸透膜,分子量約6000以上の物質を除去できる限外ろ過膜,又はこれらの膜を組み合わせた製造システムにより水を精製する方法)により製する場合,微生物による製造システムの汚染に特に注意し,蒸留法により製したものと同等の水質をもつものとする. |
参考情報 製薬用水の品質管理〈GZ-2-172〉 1.4. 注射用水 「注射用水」及び「注射用水(容器入り)」の規格及び試験方法は,日本薬局方医薬品各条で規定されている. 「注射用水」は,「常水」にイオン交換,逆浸透等による適切な前処理を行った水又は「精製水」の,蒸留又は超ろ過(RO/UF:Reverse Osmosis and/or Ultrafiltration)により製造する.蒸留法により製造する場合,飛沫同伴による汚染が起こらないように留意する.超ろ過法により製造する場合,長期間にわたるバリデーションと綿密な日常管理により,蒸留法により製造した水と同等の品質の水が恒常的に製造されることが保証される必要がある. |
(続き) 逆浸透膜又は限外ろ過膜を単独であるいは組み合わせて用いた注射用水製造システムのいずれにおいても,注射用水に適した水が安定して製造されることが,前処理装置を含む製造システム全体によって保証されることが肝要である。 超ろ過法による製造システムに関しては,水質分析,計器によるモニタリング及び透過水量監視等の日常管理を行うとともに,定期的な膜の外観検査及びエアリーク試験を実施し,併せて使用済みの膜の引張り強度,リークの有無や程度について試験を行って膜の劣化の度合いを診断し,膜交換の指標あるいは膜の破断の予知方法とするなど,膜の管理手法を確立しておくことが望ましい.また,これらに加えて,膜の使用条件に見合った適切な交換頻度を定めておくことが望ましい. |
6 ユーティリティ 直接滅菌剤として用いられる水蒸気 6.10 注射用水( W F I ) は、適格性評価の際に定められている規格に合致する水から生産し、微生物生育のリスクを最小化する方法( 例:70 ℃ を超える温度で常時循環させる) で貯蔵及び分配すること。W F I は、蒸留又は蒸留と同等の精製工程で生産すること。これには、電気脱脱イオン、限外濾過又はナノフィルタ処理等の他の適切な技術と組み合わせた逆浸透法が含まれ得る。 |
6 Utilities WATER SYSTEMS 6.10 Water for injections (WFI) should be produced from water meeting specifications that have been defined during the qualification process, stored and distributed in a manner which minimizes the risk of microbial growth (e.g. by constant circulation at a temperature above 70°C). WFI should be produced by distillation or by a purification process that is equivalent to distillation. This may include reverse osmosis coupled with other appropriate techniques such as electrodeionization (EDI), ultrafiltration or nanofiltration. |
6 Utilities Water systems 6.10 Water for injections (WFI) should be produced from water meeting specifications that have been defined during the qualification process, stored and distributed in a manner which minimizes the risk of microbial growth (e.g. by constant circulation at a temperature above 70°C). WFI should be produced by distillation or by a purification process that is equivalent to distillation. This may include reverse osmosis coupled with other appropriate techniques such as electrodeionization (EDI), ultrafiltration or nanofiltration. |
6 Utilities Water systems 6.10 Water for injection (WFI)) should be produced from water meeting specifications that have been defined during the qualification process, stored and distributed in a manner that minimizes the risk of microbial growth (for example, by constant circulation at a temperature above 70 °C). WFI should be produced by distillation or other suitable means. These may include reverse osmosis coupled with other appropriate techniques such as electrodeionization (EDI), ultrafiltration or nanofiltration. |
| 概 要 | 対象:注射用水 以下の要件があります。 ・蒸留又は超ろ過により製したものであること。 ・膜法の留意事項について。 |
対象:注射用水 以下の要件があります。 ・注射用水は、蒸留生産すること、または超ろ過(RO/UF:Reverse Osmosis and/or Ultrafiltration)により製造すること。 ・膜法の留意事項について。 |
対象:注射用水 以下の要件があります。 ・膜法の留意事項について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・注射用水は、蒸留生産すること、または、蒸留と同等の精製工程で生産すること。 ・蒸留と同等の精製工程には、電気脱イオン、限外濾過又はナノフィルタ処理等の他の適切な技術と組み合わせた逆浸透法が含まれ得る。 【誤記訂正】厚生労働省通知の和訳ではは”電気脱脱イオン”と記載されていますが、PIC/Sの原文は”electrodeionization”であるため、誤記です。表は正しい”電気脱イオン”を記載。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 製造用水 6.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 製造用水 6.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 製造用水 6.10と同じ内容となります。 |
| 国 | 日本 | 日本 | ― | 欧州 | |
|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | |
| 分 類 | 厚生労働省医薬食品局監視指導・麻薬対策課 | 厚生労働省医薬食品局監視指導・麻薬対策課 | GMP | GMP | |
| 法規類 タイトル |
医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン | 「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 11 COMPUTERISED SYSTEMS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 11: Computerised Systems |
|
| 発行年 | 2010年 | 2013年 | 2023年 | 2011年 | |
| 記載内容 | 別紙2 「カテゴリ分類表と対応例」 カテゴリー3 (構成設定していないソフトウエア)商業ベースで販売されている既製のパッケージソフトウェアで、それ自体は業務プロセスに合わせて構成設定していないもの (実行時のパラメータの入力のみで調整されるアプリケーション等は本カテゴリに含まれる) 備考欄「設備に合わせて仕様の設定及び機能の検証を行うことで差し支えない。単純なシステムに関しては校正で代用することも可」 |
本ガイドラインの対象外 ・ 製造記録の作成や出荷判定等のGQP省令及びGMP省令に係る業務に使用されない市販のワープロソフト、表計算ソフト等で、社会一般で広く利用されているパッケージソフトウエア及びPC。なお、それらソフトにより製造記録の作成や出荷判定等のGQP省令及びGMP省令に係業務に使用する場合は、本ガイドラインの対象とせず、バージョン番号、PCの機種番号、製造番号の記録等をシステム台帳登録することで良い。 |
3. 供給者とサービスプロバイダ 3.3 市販の製品に関する文書は、ユーザーの要求事項を満たすことを確認するために規制を受けるユーザーが照査すること。 用語 市販のソフトウェア:商業的に入手できるソフトウェア。使用適合性は広範囲のユーザーに立証される。 |
3. Suppliers and Service Providers 3.3 Documentation supplied with commercial off-the-shelf products should be reviewed by regulated users to check that user requirements are fulfilled. GLOSSARY Commercial off-the-shelf software Software commercially available, whose fitness for use is demonstrated by a broad spectrum of users. |
3.Suppliers and Service Providers 3.3 Documentation supplied with commercial off-the-shelf products should be reviewed by regulated users to check that user requirements are fulfilled. Glossary Commercial of the shelf software: Software commercially available, whose fitness for use is demonstrated by by a broad spectrum users. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・本文に”COTS”あるいは”OTS”についての記述はありません。上記のように別紙2表中に商業ベースで販売されている既製のパッケージソフトウェアについての記述があります。 |
ー | 対象医薬品:全般 以下の要件があります。 ・製薬企業は、COTSに添付のドキュメントをレビューし、使用するCOTSが要求仕様を満たすものであることを確認する必要がある、とされています。 ”COTS” についての用語の定義が記載されています。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 3. 供給者とサービスプロバイダ 3.3 用語と同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 3. 供給者とサービスプロバイダ 3.3 用語と同じ内容となります。 |
| 国 | 日本 | 米国 | 米国 | |
|---|---|---|---|---|
| 発行機関 | 厚生労働省 | U.S. Food and Drug Administration | CDER、CBER、他 | |
| 分 類 | 厚生労働省医薬食品局長通知 | Code of Federal Regulations | Guidance | |
| 法規類 タイトル |
医薬品等の承認又は許可等に係る申請等に関する電磁的記録・電子署名利用のための指針 | TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER A-General Part 11 Electronic Records; Electronic Signatures |
Guidance for Industry Part 11, Electronic Records; Electronic Signatures - Scope and Application | |
| 発行年 | 2005年 | ー | 2003年 | |
| 記載内容 | ー (レガシーシステムに関する記載なし) | ー (レガシーシステムに関する記載なし) | C. Approach to Specific Part11 Requirements 3. Legacy Systems The Agency intends to exercise enforcement discretion with respect to all part 11 requirements for systems that otherwise were operational prior to August 20, 1997, the effective date of part 11, under the circumstances specified below. This means that the Agency does not intend to take enforcement action to enforce compliance with any part 11 requirements if all the following criteria are met for a specific system: ・The system was operational before the effective date. ・The system met all applicable predicate rule requirements before the effective date. |
・The system currently meets all applicable predicate rule requirements. ・You have documented evidence and justification that the system is fit for its intended use (including having an acceptable level of record security and integrity, if applicable). If a system has been changed since August 20, 1997, and if the changes would prevent the system from meeting predicate rule requirements, Part 11 controls should be applied to Part 11 records and signatures pursuant to the enforcement policy expressed in this guidance. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・本指針は、原則として平成17年4月1日以降に提出又は保管される資料について適用する、としています。 |
対象医薬品:全般 以下の要件があります。 ・本規則は1997年8月20日以降有効、としています。 |
対象医薬品:全般 以下の要件があります。 ・システムが下記の全ての項目を満たしていればPart11を必須とはしない。 ・1997年8月20日より前に稼動していること。 ・上記有効日前の当局規制要求(適用される場合)に全て合致していたこと。 ・および現在も合致していること。 |
対象医薬品:全般 以下の要件があります。 ・システムが下記の全ての項目を満たしていればPart11を必須とはしない。 ・システムが使用目的に適合していることを示す証拠/正当化文書を有すること。 なお、part11の有効日前に稼動していたシステム(systems already in operation before the effective date of part11)をレガシーシステムと定義しています。 |
| 国 | 日本 | 日本 | 日本 | ー | ー | 欧州 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | PIC/S | EUROPEAN COMMISSION | |||||
| 分 類 | 厚生労働省医薬食品局監視指導・麻薬対策課 | 厚生労働省医薬食品局監視指導・麻薬対策課 | 厚生労働省医薬食品局監視指導・麻薬対策課 | GMP | Guidance | GMP | |||||
| 法規類 タイトル |
医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン | 医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドラインに関する質疑応答集(Q&A)について | 「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 11 MANUFACTURE OF STERILE COMPUTERISED SYSTEMS |
GOOD PRACTICES FOR COMPUTERISED SYSTEMS IN REGULATED "GXP" ENVIRONMENTS | EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 11: Computerised Systems |
|||||
| 発行年 | 2010年 | 2010年 | 2013年 | 2023年 | 2007年 | 2011年 | |||||
| 記載内容 | 別紙2 カテゴリ3、製造設備、分析機器、製造支援設備等に搭載されるシステム →バリデーション計画書・報告書の策定の対象としている。 同備考欄 設備に合わせて仕様の設定及び機能の検証を行うことで差支えない。単純なシステムに関しては校正で代用することも可 |
問12 使用目的が限定され、そのためのプログラムがハードウェア(コンピュータにより制御される機器及び設備を含む。) の供給者によって汎用機能として固定され、パラメータを設定することによって機能が実現されるシステムとして、従来ファームウェアやPLC(Programmable logic controller)として分類されていたシステムもガイドラインの適用の対象になるか。 回答12 ファームウェアやPLCであっても、このガイドライン別紙2「カテゴリ分類表と対応例」に従いシステムのリスクに応じた対応を行うこととなる。 |
問36 次の機能を有するコンピュータを搭載したPTP分包機の場合はどのカテゴリが適用されるか。 ・ PTPシート中の錠剤の抜けを光学的センサーの出力を受けて判断し、当該シートを後工程で排除する。 ・ 光学的センサーの出力を錠剤面積比として計算し、一定以上の欠損錠を不良錠として判断する。 ・ 製造シート(良品、不良品)枚数、製造数量等の管理上のデータ処理をする。 なお、駆動部の制御はPLC(Programmable logic controller)にて行う。 |
(続き)回答36 このPTP分包機が単体として使用されている場合であって、単に運転条件の設定のみで機能が実現されるものである場合にはガイドラインのカテゴリ3に分類される。このため、設備の適格性の確認においてプログラムの機能が全て確認できる場合は設備のバリデーションに含めて実施することで差し支えない。ただし、これらの単体設備や機器等をさらに上位のコンピュータに接続するためにインターフェース等をあらためて作成し、上位コンピュータから制御して使用している場合には、原則としてガイドラインのカテゴリ5が適用される。 |
問46 このガイドラインで新たに適用の範囲とされたファームウェアやPLC等の既存のシステムについては、どの様な取り扱いが必要となるか。 回答46 既存のシステムについては当該設備としてのバリデーションが適切に実施されている場合は、コンピュータ化システムもバリデートされたものとみなしても差し支えない。ただし、当該設備において、システム上の障害の発生が考えられる場合は、改めてバリデーションを実施する必要がある。 |
原則 本文書はGMP規制をうける業務の一部として使用されるコンピュータ化システム全形態に適用する。コンピュータ化システムはソフトウェア及びハードウェアの構成要素が一体となって特定の機能を満たすものである。 |
PRINCIPLE This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfil certain functionalities. |
8. MANAGEMENT AND RESPONSIBILITIES 8.5 Within the regulated user organisation there should be clearly defined responsibilities for the management of all ICT products, computerised systems and projects. Management should cover the full spectrum, from simple input/output devices and programmable logic controllers (PLCs) through to integrated supervisory or information systems and business management levels. These responsibilities should involve development and administration of policies on purchase of IT products, as well as the introduction, commissioning and maintenance of IT products. The responsibilities should extend to development and implementation of formal monitoring, auditing and servicing of each system and designate the related documentation and records for such activities. |
13. TESTING 13.3 For some simpler GxP systems, for example certain PLCs and systems based on basic algorithms or logic sets, the functional testing may provide adequate assurance of reliability of the computerised system. For critical and/or more complex systems the verification testing that is conducted at the IQ, OQ & PQ stages provides only a limited level of assurance that the system does what it purports to do, reliably. This level of testing provides only limited assurance of the operation and reliability of hidden functions and code. For complex systems there should also be a high level of assurance that the development of the software has ensured delivery and operation of a quality product that is structurally sound, clearly defined and controlled. |
27. GLOSSARY OF TERMS Embedded System A system, usually microprocessor of PLC based, whose sole purpose is to control a particular piece of automated equipment. This is contrasted with a standalone computer system. 28.ABBREVEATIONS USED IN THE DOCUMENT PLC: Programmable Logic Controller |
Principle This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill certain functionalities. |
| 概 要 | 対象医薬品:全般 ・PLCについて直接の記述はありません。製造装置等に組み込まれたPLCはカテゴリ3に含まれます。ただし、ユーザがPLC上でアプリケーションを作成した場合には、カテゴリの分類はカテゴリ4または5になります。 |
対象医薬品:全般 以下の要件があります。 ・PLCのバリデーションについて、基本的な考え方の記述があります。 |
対象医薬品:全般 以下の要件があります。 ・PLCのバリデーションについて、基本的な考え方の記述があります。 |
対象医薬品:全般 以下の要件があります。 ・PLCのバリデーションについて、基本的な考え方の記述があります。 |
対象医薬品:全般 以下の要件があります。 ・PLCのバリデーションについて、基本的な考え方の記述があります。 |
対象医薬品:全般 以下の要件があります。 ・GMPの規制を受けるコンピュータシステムにPLCが含まれる場合、PLCシステムはバリデーションの対象となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 原則と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・管理対象とするコンピュータシステムの範囲を単純な入出力機器あるいはPLCからインテグレーションされた情報システムまでとする旨、記述があります。 なお、原文中のITおよびICTは、各々、Information Technology、Information and Commmunications Technologyの略です。 |
対象医薬品:全般 以下の要件があります。 ・PLCで構成される簡単なGxPシステムの場合、機能テストにてコンピュータの信頼性を保証することで良い、としています。 |
対象医薬品:全般 以下の要件があります。 ・GLOSSARY にPLCで構成されるEmbedded Systemの記述があります。 ABBREVEATIONにPLCの記述があります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 原則と同じ内容となります。 |
| 国 | 日本 | 日本 | 米国 | 米国 | ー | 欧州 | ー | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CBER | PIC/S | EUROPEAN COMMISSION | WHO | ||||||||||||||
| 分 類 | 厚生労働省医薬食品局監視指導・麻薬対策課 | 厚生労働省・生活衛生局監視指導・麻薬対策課 事務連絡 | Code of Federal Regulations | Final Guidance for Industry and FDA Staff | GMP | GMP | GMP | ||||||||||||||
| 法規類 タイトル |
医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(10) PIC/S GMPガイドライン アネックス11 コンピュータ化システム |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER A--GENERAL PART 11 ELECTRONIC RECORDS; ELECTRONIC SIGNATURES |
FDA General Principles of Software Validation | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 11 MANUFACTURE OF STERILE COMPUTERISED SYSTEMS |
EU Guidelines to Good Manufacturing Practice Medical Products for Human and Veterinary Use Annex 11 Computerized Systems |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 3 Good manufacturing practices: guidelines on validation Appendix 5. Validation of computerized systems WHO Technical Report Series, No. 1019, 2019 |
||||||||||||||
| 発行年 | 2010年 | 2017年 | --- | 2002年 | 2023年 | 2011年 | 2019年 | ||||||||||||||
| 記載内容 | 1. 総則 1.2 コンピュータ化システムの取扱い (前略)また、このガイドラインの対象となるコンピュータ化システムで「医薬品等の承認又は許可等に係る申請等に関する電磁的記録・電子署名利用のための指針」(平成17年4月1日薬食発第0401022号)及び「薬事法及び採血及び供血あつせん業取締法の一部を改正する法律の施行に伴う医薬品,医療機器等の製造管理及び品質管理(GMP/QMS)に係る省令及び告示の制定及び改廃について」 (平成17年3月30日薬食監麻発第0330001号)第3章第3 35.「その他(電磁的記録等について)」の適用を受けるコンピュータ化システムは、併せてそれら要件を備える必要がある。 |
運用段階 7. データの保存 7.1 データは、物理学的方法及び電子的方法によって損傷害から守ること。記録されたデータはアクセスのしやすさ、可読性、正確性を確認すること。 7.2 すべての該当データの定期的なバックアップを行うこと。バックアップデータの完全性と正確性及びデータを保存する能力は、バリデーションで確認し、定期的にモニターすること。 |
Subpart B-Electronic Records Sec. 11.10 Controls for closed systems. Persons who use closed systems to create, modify, maintain, or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity, and, when appropriate, the confidentiality of electronic records, and to ensure that the signer cannot readily repudiate the signed record as not genuine. Such procedures and controls shall include the following: (a) Validation of systems to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records. |
(b) The ability to generate accurate and complete copies of records in both human readable and electronic form suitable for inspection, review, and copying by the agency. Persons should contact the agency if there are any questions regarding the ability of the agency to perform such review and copying of the electronic records. (c) Protection of records to enable their accurate and ready retrieval throughout the records retention period. (d) Limiting system access to authorized individuals. |
(e) Use of secure, computer-generated, time-stamped audit trails to independently record the date and time of operator entries and actions that create, modify, or delete electronic records. Record changes shall not obscure previously recorded information. Such audit trail documentation shall be retained for a period at least as long as that required for the subject electronic records and shall be available for agency review and copying. (f) Use of operational system checks to enforce permitted sequencing of steps and events, as appropriate. |
(g) Use of authority checks to ensure that only authorized individuals can use the system, electronically sign a record, access the operation or computer system input or output device, alter a record, or perform the operation at hand. (h) Use of device (e.g., terminal) checks to determine, as appropriate, the validity of the source of data input or operational instruction. (i) Determination that persons who develop, maintain, or use electronic record/electronic signature systems have the education, training, and experience to perform their assigned tasks. |
(j) The establishment of, and adherence to, written policies that hold individuals accountable and responsible for actions initiated under their electronic signatures, in order to deter record and signature falsification. | Subpart B-Electronic Records §11.30 Controls for open systems. Persons who use open systems to create, modify, maintain, or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity, and, as appropriate, the confidentiality of electronic records from the point of their creation to the point of their receipt. Such procedures and controls shall include those identified in §11.10, as appropriate, and additional measures such as document encryption and use of appropriate digital signature standards to ensure, as necessary under the circumstances, record authenticity, integrity, and confidentiality. |
SECTION 2. SCOPE 2.4. REGULATORY REQUIREMENTS FOR SOFTWARE VALIDATION In addition, computer systems used to create, modify, and maintain electronic records and to manage electronic signatures are also subject to the validation requirements.(See 21 CFR §11.10(a).) |
OPERATIONAL PHASE 7. Data Storage 7.1 Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability and accuracy. Access to data should be ensured throughout the retention period. 7.2 Regular back-ups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically. |
Operational Phase 7. Data Storage 7.1 Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability and accuracy. Access to data should be ensured throughout the retention period. 7.2 Regular back-ups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically. |
2. GLOSSARY audit trail. The audit trail is a form of metadata that contains information associated with actions that relate to the creation, modification or deletion of GXP records. An audit trail provides for secure recording of lifecycle details, such as creation, additions, deletions or alterations of information in a record, either paper or electronic, without obscuring or overwriting the original record. An audit trail facilitates the reconstruction of the history of such events relating to the record, regardless of its medium, including the “who”, “what”, “when” and “why” of the action. For example, in a paper record, an audit trail of a change would be documented via a single-line cross-out that allows the original entry to remain legible and documents the initials of the person making the change, |
(続き) the date of the change and the reason for the change, as required to substantiate and justify the change. In electronic records, secure, computer-generated, time stamped audit trails should allow for reconstruction of the course of events relating to the creation, modification and deletion of electronic data. Computer-generated audit trails should retain the original entry and document the user identification and the time/date stamp of the action, as well as the reason for the change, as required to substantiate and justify the action. Computer-generated audit trails may include discrete event logs, history files, database queries or reports, or other mechanisms that display events related to the computerized system, specific electronic records or specific data contained within the record. |
(続き) backup. A backup means a copy of one or more electronic files created as an alternative in case the original data or system are lost or become unusable (e.g. in the event of a system crash or corruption of a disk). It is important to note that backup differs from archiving, in that backup copies of electronic records are typically only temporarily stored for the purposes of disaster recovery and may be periodically overwritten. Such temporary backup copies should not be relied upon as an archiving mechanism. |
(続き) data. All original records and true copies of original records, including source data and metadata, and all subsequent transformations and reports of these data, which are generated or recorded at the time of the GMP activity and allow full and complete reconstruction and evaluation of the GMP activity. Data should be accurately recorded by permanent means at the time of the activity. Data may be contained in paper records (such as worksheets and logbooks), electronic records and audit trails, photographs, microfilm or microfiche, audio or video files or any other media whereby information related to GMP activities is recorded. |
5. Requirements specifications User requirements specifications 5.3 Other aspects to be included in the URS may include, but are not limited to: (略) ■controls that assure that all steps that create, modify or delete electronic data related to GMP purposes will be recorded in independent, computer-generated audit trails or other metadata, or alternate documents that record the “what” (e.g. original entry), “who” (e.g. user ID), “when” (e.g. time/date stamp) and “why” (e.g. reason) of the action; |
(続き) ■ the ability to archive and retrieve the electronic data in a manner that assures that the archive copy preserves the full content of the original electronic data set, including all metadata needed to fully reconstruct the GMP activity. The archive copy should also preserve the meaning of the original electronic data set; ■ input/output checks, including implementation of procedures for the review of original electronic data and metadata, such as audit trails; (略) |
11. Standard operating procedures and training 11.2 Procedures and training programmes that should be developed include, but are not necessarily limited to: ■system use procedures that address: (略) - review of the electronic data and associated metadata (such as audit trails) and how the source electronic records will be reconciled with printouts, if any; (略) |
12. Performance qualification and user acceptance testing 12.5 UAT should be conducted by system users, to verify the adequacy of the system, use of SOPs and training programmes. The UAT should include verification of the ability to generate and process only valid data within the computerized system, including the ability to efficiently review electronic data and metadata, such as audit trails. SOPs should be finalized and approved upon completion of performance qualification. | 13. System operation and maintenance Operation and maintenance 13.11 There should be written procedures governing system operation and maintenance, including, for example: (略) ■ssystem use and review of electronic data and metadata (such as audit trails); (略) Data migration 13.13 Where electronic data are transferred from one system to another, itshould be demonstrated that data are not altered during the migration process. Conversion of data to a different format should be considered as data migration. Where data are transferred to another medium, they must be verified as an exact copy, prior to any destruction of the original data. |
14. System retirement 14.3 Records should be archived in a readable form and in a manner that preserves the accessibility, readability and integrity of the data of the source electronic records throughout the required records retention period. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・”電磁的記録”との表記がされています。具体的な内容について記述はありません。関連資料を適用する旨記述があります。 |
対象医薬品:全般 以下の要件があります。 ・運用段階におけるデータの保存についての要件が記述されています。 |
対象医薬品:全般 以下の要件があります。 ・クローズドシステム(システムアクセスが電子記録内容の責任者によって管理されている場合)についての手順と管理方法が示されています。 (バリデーション) |
対象医薬品:全般 以下の要件があります。 ・クローズドシステム(システムアクセスが電子記録内容の責任者によって管理されている場合)についての手順と管理方法が示されています。 (正確で完全な記録、記録の保護、記録へのアクセス) |
対象医薬品:全般 以下の要件があります。 ・クローズドシステム(システムアクセスが電子記録内容の責任者によって管理されている場合)についての手順と管理方法が示されています。 (監査証跡、システムチェック) |
対象医薬品:全般 以下の要件があります。 ・クローズドシステム(システムアクセスが電子記録内容の責任者によって管理されている場合)についての手順と管理方法が示されています。 (権限、デバイスチェック、教育訓練) |
対象医薬品:全般 以下の要件があります。 ・クローズドシステム(システムアクセスが電子記録内容の責任者によって管理されている場合)についての手順と管理方法が示されています。 (電子署名) |
対象医薬品:全般 以下の要件があります。 ・オープンシステムが電子記録内容の責任者によって管理されている場合についての手順と管理方法が示されています。クローズドシステムの要件に加えて付加的な要件が必要とされています。 |
対象医薬品:全般 以下の要件があります。 ・ERはバリデーションの対象の旨記述があります。21 CFR Part 11§11.10(a)に基づいて検証を行うこととされています。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 運用段階 7. データの保存 7.1、7.2と同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 運用段階 7. データの保存 7.1、7.2と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・Glossaryにおける ”audit trail”に Electronic data を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 Glossaryにおける ”audit trail”に Electronic data を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 ・Glossaryにおける ”backup”に Electronic data を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 ・Glossaryにおける”data”に Electronic data を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 ・要求仕様においてElectronic data の取り扱いについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・要求仕様においてElectronic data の取り扱いについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・SOPと訓練においてElectronic data の取り扱いについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・PQとUser acceptance testingにおいてElectronic data の取り扱いについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・システム操作と保守においてElectronic data の取り扱いについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・システムの廃棄においてElectronic data の取り扱いについての記述があります。 |
| 国 | 日本 | 日本 | 米国 | 米国 | 米国 | ー | 欧州 | ー | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | U.S. Food and Drug Administration | CBER | PIC/S | EUROPEAN COMMISSION | WHO | |||
| 分 類 | 厚生労働省医薬品食品局監視指導・麻薬対策課 | 厚生労働省・生活衛生局監視指導・麻薬対策課 事務連絡 | Code of Federal Regulations | Code of Federal Regulations | Final Guidance for Industry and FDA Staff | GMP | GMP | GMP | |||
| 法規類 タイトル |
医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(10) PIC/S GMPガイドライン アネックス11 コンピュータ化システム |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER A--GENERAL PART 11 ELECTRONIC RECORDS; ELECTRONIC SIGNATURES |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER A--GENERAL PART 11 ELECTRONIC RECORDS; ELECTRONIC SIGNATURES |
FDA General Principles of Software Validation | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 11 MANUFACTURE OF STERILE COMPUTERISED SYSTEMS |
EU Guidelines to Good Manufacturing Practice Medical Products for Human and Veterinary Use Annex 11 Computerized Systems |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 3 Good manufacturing practices: guidelines on validation Appendix 5. Validation of computerized systems WHO Technical Report Series, No. 1019, 2019 |
|||
| 発行年 | 2010年 | 2017年 | ー | ー | 2002年 | 2023年 | 2011年 | 2019年 | |||
| 記載内容 | 1. 総則 1.2 コンピュータ化システムの取扱い (前略)また、このガイドラインの対象となるコンピュータ化システムで「医薬品等の承認又は許可等に係る申請等に関する電磁的記録・電子署名利用のための指針」(平成17年4月1日薬食発第0401022号)及び「薬事法及び採血及び供血あつせん業取締法の一部を改正する法律の施行に伴う医薬品,医療機器等の製造管理及び品質管理(GMP/QMS)に係る省令及び告示の制定及び改廃について」 (平成17年3月30日薬食監麻発第0330001号)第3章第3 35.「その他(電磁的記録等について)」の適用を受けるコンピュータ化システムは、併せてそれら要件を備える必要がある。 |
運用段階 14. 電子署名 a. 会社内での手書きの署名と同じ効力がある b. 記録が存在する限り、個々の記録と関連付ける c. 署名を行った日時を含む |
Subpart A-General Provisions §11.3 Definitions. (7) Electronic signature means a computer data compilation of any symbol or series of symbols executed, adopted, or authorized by an individual to be the legally binding equivalent of the individual's handwritten signature |
Subpart B-Electronic Records §11.200 Electronic signature components and controls. (a) Electronic signatures that are not based upon biometrics shall: (1) Employ at least two distinct identification components such as an identification code and password. (i) When an individual executes a series of signings during a single, continuous period of controlled system access, the first signing shall be executed using all electronic signature components; subsequent signings shall be executed using at least one electronic signature component that is only executable by, and designed to be used only by, the individual. |
(続き) (ii) When an individual executes one or more signings not performed during a single, continuous period of controlled system access, each signing shall be executed using all of the electronic signature components. (2) Be used only by their genuine owners; and (3) Be administered and executed to ensure that attempted use of an individual's electronic signature by anyone other than its genuine owner requires collaboration of two or more individuals. (b) Electronic signatures based upon biometrics shall be designed to ensure that they cannot be used by anyone other than their genuine owners. |
SECTION 2. SCOPE 2.4. REGULATORY REQUIREMENTS FOR SOFTWARE VALIDATION In addition, computer systems used to create, modify, and maintain electronic records and to manage electronic signatures are also subject to the validation requirements.(See 21 CFR §11.10(a).) |
OPERATIONAL PHASE 14. Electronic Signature Electronic records may be signed electronically. Electronic signatures are expected to: a. have the same impact as hand-written signatures within the boundaries of the company, b. be permanently linked to their respective record, c. include the time and date that they were applied. |
Operational Phase 14. Electronic Signature Electronic records may be signed electronically. Electronic signatures are expected to: a. have the same impact as hand-written signatures within the boundaries of the company, b. be permanently linked to their respective record, c. include the time and date that they were applied. |
2. Glossary archiving. Archiving is the process of protecting records from the possibility of being further altered or deleted, and storing these records under the control of independent data management personnel throughout the required retention period. Archived records should include, for example, associated metadata and electronic signatures. |
5. Requirements specifications User requirements specifications 5.3 Other aspects to be included in the URS may include, but are not limited to: (略) ■electronic signatures; (略) 11. Standard operating procedures and training 11.2 Procedures and training programmes that should be developed include, but are not necessarily limited to: (略) ■system use procedures that address: - mechanisms for signing electronic data; (略) |
13. System operation and maintenance Data migration 13.14 Procedures for data migration may vary greatly in complexity, and measures to ensure appropriate transfer of data should be commensurate with identified risks. Migrated data should remain usable and should retain their content and meaning. The value and/or meaning of and links between a system audit trail and electronic signatures should be ensured in a migration process. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・具体的な内容について記述はありません。関連資料を適用する旨記述があります。 |
対象医薬品:全般 以下の要件があります。 ・運用段階における電子署名についての要件が記述されています。 |
対象医薬品:全般 以下の要件があります。 ・電子署名についての定義が示されています。 |
対象医薬品:全般 以下の要件があります。 ・電子署名についての要件が記述されています。(バイオメトリクスに基づかない署名、バイオメトリクスに基づいた署名の注意事項があります) |
対象医薬品:全般 以下の要件があります。 ・電子署名についての要件が記述されています。(バイオメトリクスに基づかない署名、バイオメトリクスに基づいた署名の注意事項があります) |
対象医薬品:全般 以下の要件があります。 ・ER/ESはバリデーションの対象の旨記述があります。21 CFR Part 11§11.10(a)に基づいて検証を行うこととされています。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 運用段階 14 電子署名 a. 、b.、c.と同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 運用段階 14 電子署名 a. 、b.、c.と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・Glossaryにおいて ”archiving” に electronic signature を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 ・要求仕様、SOPと訓練においてelectronic signature についての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・システムの廃棄においてelectronic signature についての記述があります。 |
| 国 | 日本 | 日本 | 米国 | ー | 欧州 | ー | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | CBER | PIC/S | EUROPEAN COMMISSION | WHO | |||||||||||
| 分 類 | 厚生労働省医薬品食品局監視指導・麻薬対策課 | 厚生労働省・生活衛生局監視指導・麻薬対策課 事務連絡 | Final Guidance for Industry and FDA Staff | GMP | GMP | GMP | |||||||||||
| 法規類 タイトル |
医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(10) PIC/S GMPガイドライン アネックス11 コンピュータ化システム |
FDA General Principles of Software Validation | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 11 MANUFACTURE OF STERILE COMPUTERISED SYSTEMS |
EU Guidelines to Good Manufacturing Practice Medical Products for Human and Veterinary Use Annex 11 Computerised Systems |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 3 Good manufacturing practices: guidelines on validation Appendix 5. Validation of computerized systems WHO Technical Report Series, No. 1019, 2019 |
|||||||||||
| 発行年 | 2010年 | 2017年 | 2002年 | 2023年 | 2011年 | 2019年 | |||||||||||
| 記載内容 | 1. 総則 1.1 目的 (前略)このガイドラインにおいては、コンピュータ化システムの開発から、検証、運用管理及び廃棄までの流れを総合してコンピュータ化システムのライフサイクルという。ライフサイクル全体の構成を別紙1「コンピュータ化システムのライフサイクルモデル」に示す。 |
一般事項 1.リスクマネジメント リスクマネジメントは、患者の安全性、データの完全性、製品の品質を考慮に入れ、コンピュータ化システムのライフサイクル全体に適用すること。リスクマネジメントの一部として、バリデーションの範囲とデータの完全性の判断は正当化し、文書化したコンピュータ化システムのリスク評価に基づいて行うこと。 |
開発・検証段階 4.バリデーション 4.1 バリデーションの文書及び報告書はライフサイクルの該当する段階を網羅すること。製造業者は、リスク評価を基にした、基準、プロトコル、許容基準、手順書、記録を正当化できるようにすること。 4.4 要求事項仕様書は、コンピュータ化システムに要求された機能を記述し、文書化されたリスク評価及びGMPへの影響に基づいてこと。ユーザー要求事項は、ライフサイクルを通じて追跡可能であること。 4.6 特注或いはカスタマイズされたコンピュータ化システムのバリデーションについては、システムの全ライフサイクルを通じて品質及び性質について採られた措置は正式に評価して報告を保証するための工程があること。 |
用語 ライフサイクル:設計、規格、プログラム作成、試験、設置、操作、保守管理を含めた、初期の要求事項から廃棄までのシステムの耐用期間における全段階。 |
SECTION 4. PRINCIPLES OF SOFTWARE VALIDATION This section lists the general principles that should be considered for the validation of software. 4.4. SOFTWARE LIFE CYCLE Software validation takes place within the environment of an established software life cycle. The software life cycle contains software engineering tasks and documentation necessary to support the software validation effort. In addition, the software life cycle contains specific verification and validation tasks that are appropriate for the intended use of the software. This guidance does not recommend any particular life cycle models - only that they should be selected and used for a software development project. |
5.1. SOFTWARE LIFE CYCLE ACTIVITIES This guidance does not recommend the use of any specific software life cycle model. Software developers should establish a software life cycle model that is appropriate for their product and organization. The software life cycle model that is selected should cover the software from its birth to its retirement. Activities in a typical software life cycle model include the following: ・Quality Planning ・System Requirements Definition ・Detailed Software Requirements Specification ・Software Design Specification ・Construction or Coding ・Testing ・Installation ・Operation and Support ・ Maintenance ・Retirement |
(続き) Several software life cycle models (e.g., waterfall, spiral, rapid prototyping, incremental development, etc.) are defined in FDA’s Glossary of Computerized System and Software Development Terminology, dated August 1995. These and many other life cycle models are described in various references listed in Appendix A.G9:G10 |
GENERAL 1. Risk Management Risk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. (略) |
PROJECT PHASE 4. Validation 4.1 The validation documentation and reports should cover the relevant steps of the life cycle. (略) 4.4 (略) User requirements should be traceable throughout the life-cycle. 4.6 For the validation of bespoke or customised computerised systems there should be a process in place that ensures the formal assessment and reporting of quality and performance measures for all the life-cycle stages of the system. |
GLOSSARY Life cycle All phases in the life of the system from initial requirements until retirement including design, specification, programming, testing, installation, operation, and maintenance. |
GENERAL 1. Risk Management Risk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. (略) |
PROJECT PHASE 4. Validation 4.1 The validation documentation and reports should cover the relevant steps of the life cycle. (略) 4.4 (略) User requirements should be traceable throughout the life-cycle. 4.6 For the validation of bespoke or customised computerised systems there should be a process in place that ensures the formal assessment and reporting of quality and performance measures for all the life-cycle stages of the system. |
Glossary: Life cycle: All phases in the life of the system from initial requirements until retirement including design, specification, programming, testing, installation, operation, and maintenance. |
1. Introduction and scope 1.4 Computerized systems should be maintained throughout the system lifecycle, in a validated state, with risk-based controls for the operational phase, which may include, but are not limited to, system planning; preparation and verification of standard operating procedures (SOPs) and training programmes; system operation and maintenance, including handling of software and hardware updates; monitoring and review; change management; and incident reporting, followed by system retirement. |
2. Glossary audit trail. The audit trail is a form of metadata that contains information associated with actions that relate to the creation, modification or deletion of GXP records. An audit trail provides for secure recording of lifecycle details, such as creation, additions, deletions or alterations of information in a record, either paper or electronic, without obscuring or overwriting the original record. (略) commercial off-the-shelf software (COTS). A vendor-supplied software component of a computerized system for which the user cannot claim complete control of the software life-cycle. |
(続き) system life-cycle. The period of time that starts when a computerized system is conceived and ends when the system is retired and decommissioned, taking into consideration regulatory requirements. The system life-cycle typically includes a planning phase; a development phase that includes a design phase and a programming and testing phase; a qualification and release phase that includes a system integration and testing phase; a validation phase; a release phase; an operation and maintenance phase; and, finally, a system retirement phase. |
14. System retirement 14.1 System retirement should be considered as a system life-cycle phase. It should be planned, risk based and documented. If migration or archiving of GMP-relevant data (1, 2) is necessary, the process must be documented. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・コンピュータ化システムの開発から、検証、運用管理及び廃棄までの流れをライフサイクルとして規定してます。具体的な記述は同ガイドライン別紙1に”コンピュータ化システムのライフサイクルモデル”として記述があります。 |
対象医薬品:全般 以下の要件があります。 ・リスクマネジメントは、、コンピュータ化システムのライフサイクル全体に適用されること。 具体的な内容について記述はありません。 |
対象医薬品:全般 以下の要件があります。 ・バリデーションの文書及び報告書はライフサイクルの該当する段階を網羅すること。 ・ユーザー要求事項は、ライフサイクルを通じて追跡可能であること。 ・カスタマイズされたコンピュータ化システムについての留意事項が示されています。 |
対象医薬品:全般 以下の要件があります。 ・ライフサイクルについての定義が示されています。 |
対象医薬品:全般 以下の要件があります。 ・バリデーションはライフサイクルの各要素に対して実施すべきとの記述があります。またソフトウエアの開発のためにのみ採用されるライフサイクルモデルはバリデーションに適当ではないとの記述があります。 ・ライフサイクルについて典型的なものを例に具体的な記述があります。 ・ライフサイクルの各種モデルについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・ライフサイクルについてはソフトウエアライフサイクルとして記述されています。ハードウエアについてはライフサイクルに関連した記述はありません。 |
対象医薬品:全般 以下の要件があります。 ・ライフサイクルについてはソフトウエアライフサイクルとして記述されています。ハードウエアについてはライフサイクルに関連した記述はありません。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 1.リスクマネジメントと同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 4.バリデーション 4.1、4.4、4.6と同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム GLOSSARY Life cycleと同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 1.リスクマネジメントと同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 4.バリデーション 4.1、4.4、4.6と同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム GLOSSARY Life cycleと同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・INTRODUCTION AND SCOPEにコンピュータシステムのライフサイクルについての記述があります。 |
対象医薬品:全般 以下の要件があります。 ・Glossaryにおいて ”audit trail”、”COTS”に life cycle を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 ・Glossaryにおいて ”system life cycle” に life cycle を含む記述があります。 |
対象医薬品:全般 以下の要件があります。 ・システム廃棄 についての記述があります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 米国 | ー | 欧州 | ー | |
|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | EUROPEAN COMMISSION | WHO | |
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬品食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | |
| 法規類 タイトル |
薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing ― Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS Current Good Manufacturing Practice |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|
| 発行年 | 2021年 | 2011年 | 2012年 | 2025年 | 2004年 | 2023年 | 2022年 | 2022年 | |
| 記載内容 | 第二章 医薬品等の製造業 第一節 医薬品の製造業 (一般区分の医薬品製造業者等の製造所の構造設備) (特定生物由来医薬品等の医薬品製造業者等の製造所の構造設備) 第八条 (中略)(以下「特定生物由来医薬品等」と総称する。)に係る製品の医薬品製造業者等の製造所の構造設備の基準は、第六条(無菌医薬品に係る製品の製造を行う場合においては、前二条)に定めるもののほか、次のとおりとする。 一 特定生物由来医薬品等に係る製品の製造所(包装、表示又は保管のみを行う製造所を除く。)は、次に定めるところに適合するものであること。 ロ 清浄区域には、排水口を設置しないこと。ただし、次に定めるところに適合する場合であつて、やむを得ないと認められるときは、この限りでない。 |
(続き) (1) 排水口は、清掃が容易なトラツプ及び排水の逆流を防止するための装置を有するものであること。 (2) トラツプは、消毒を行うことができる構造のものであること。 (3) 床の溝は、浅く清掃が容易なものであり、かつ、排水口を通じて、製造区域(培養、抽出及び精製作業、原料の秤量作業、容器の洗浄及び乾燥作業、薬剤の調製及び充填作業並びに容器の閉塞及び包装作業を行う場所並びに更衣を行う場所をいう。以下この号において同じ。)の外へ接続されていること。 ハ 無菌区域は、次に定めるところに適合するものであること。 (1) 排水口を設置しないこと。 (2) 流しを設置しないこと。 |
6 構造設備 6.1 構造設備の設計上の要点 17) 無菌操作区域には排水口や流しを設置しないこと。また、その他の支援区域のうちグレードCに排水口を設ける場合は清掃が容易で消毒ができるトラップ及び排水の逆流を防ぐ装置を有するなど排水口からの汚染防止を考慮すること。床に溝を設ける場合は浅く、清掃が容易な構造であること。 |
5. 建物および施設 5.1 構造設備の設計上の要点 16) グレードB以上の区域内には、流しや排水口を設けないこと。その他の支援区域(グレードC又はグレードD)に排水口を設ける場合は、清掃が容易で消毒ができるトラップ及び排水の逆流を防ぐ装置を有するなど排水口からの汚染防止を考慮すること。床に溝を設ける場合は、浅く、清掃が容易な構造であること。 |
4 建物 4.9 シンク及び排水口を、グレードA及びグレードBの区域内に設置しないこと。その他のクリーンルーム内では、エアブレイクが機器又はシンクと排水口の間に取り付けられていること。低い等級のクリーンルーム内の床排水口には、逆流を防止する設計のトラップ又はウォータシールが取り付けられ、定期的に清浄化され、消毒され、保守管理されていること。 |
IV. BUILDINGS AND FACILITIES E. Design With rare exception, drains are considered inappropriate for classified areas of the aseptic processing facility other than Class 100,000(ISO 8) areas. It is essential that any drain installed in an aseptic processing facility be of sutitable design. |
4 Premises 4.9 Sinks and drains should be prohibited in the grade A and grade B areas. In other cleanrooms, air breaks should be fitted between the machine or sink and the drains. Floor drains in lower grade cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be regularly cleaned, disinfected and maintained. |
4 Premises 4.9 Sinks and drains should be prohibited in the grade A and grade B areas. In other cleanrooms, air breaks should be fitted between the machine or sink and the drains. Floor drains in lower grade cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be regularly cleaned, disinfected and maintained. |
4. Premises 4.9 Sinks and drains should be prohibited in the grade A and B areas. In other cleanrooms, air breaks should be fitted between the machine or sink and the drains. Floor drains in lower-grade cleanrooms should be fitted with traps or water seals designed to prevent backflow and should be regularly cleaned, disinfected and maintained. |
| 概 要 | 対象医薬品:特定生物由来医薬品 以下の要件があります。 ・清浄区域には、排水口を設置しないこと。 |
対象医薬品:特定生物由来医薬品 以下の要件があります。 ・清浄区域に、やむを得ない排水口が認められるトラツプ、逆流防止装置、床の溝についての条件について。 ・無菌区域、排水口、流しを設置しないこと。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌操作区域では排水口、流しは、設置しないこと。 ・グレードCに排水口を設ける場合、消毒できるトラップ及び逆流防止装置を設置。 ・床に溝を設ける場合の条件について。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・グレードB以上の区域では、流し台や排水口を設置しないこと。 ・グレードCおよびDに排水口を設ける場合は、清掃が容易で消毒できるトラップ及び逆流防止装置を設置。 ・床に溝を設ける場合の条件について。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードAおよびBの区域には、流し台や排水口を設置しないこと。 ・その他のグレードのクリーンルームにおいては、エアブレイク、排水トラップまたは水封機構の設置、定期的な清浄および消毒等による保守管理を要する。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 *エアブレイクの用語について、GMP構造設備要求比較 建築 002:排水からの汚染防止2の概要欄を参照ください。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・クラス100,000 (ISO 8)を除く、無菌操作施設のクラス区分された区域に排水口は設置しないこと。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | 米国 | ー | ー | 欧州 | 欧州 | ー | ー | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CDER | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | ||
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬品食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長通知 | Code of Federal Regulations | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | ||
| 法規類 タイトル |
薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | PIC/SのGMPガイドラインを活用する際の考え方について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
原薬GMPのガイドラインについて(ICHQ7) | TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, No. 986 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||
| 発行年 | 2021年 | 2011年 | 2012年 | 2020年 | 2025年 | 2001年 | 2023年 | 2004年 | 2021年 | 2023年 | 2015年 | 2022年 | 2014年 | 2022年 | ||
| 記載内容 | 第二章 医薬品等の製造業 第一節 医薬品の製造業 (一般区分の医薬品製造業者等の製造所の構造設備) 第六条 施行規則第二十五条第一項第四号の区分及び施行規則第三十五条第一項第四号の区分の製造業者及び医薬品等外国製造業者(法第十三条の三第一項に規定する医薬品等外国製造業者をいう。)(以下「医薬品製造業者等」と総称する。)の製造所の構造設備の基準は、次のとおりとする。 5 原薬に係る製品の作業所のうち、最終の精製以後の製造工程において、最終の精製を経た中間製品を容器へ充填及び閉塞するまでの作業を行う作業室及び原薬に係る製品以外の製品の作業所のうち、原料の秤量作業、製品の調製作業、充填作業又は閉塞作業を行う作業室は、次に定めるところに適合するものであること。 ハ 室内の排水設備は、作業室の汚染を防止するために必要な構造であること。 |
(特定生物由来医薬品等の医薬品製造業者等の製造所の構造設備) 第八条 (中略)(以下「特定生物由来医薬品等」と総称する。)に係る製品の医薬品製造業者等の製造所の構造設備の基準は、第六条(無菌医薬品に係る製品の製造を行う場合においては、前二条)に定めるもののほか、次のとおりとする。 一 特定生物由来医薬品等に係る製品の製造所(包装、表示又は保管のみを行う製造所を除く。)は、次に定めるところに適合するものであること。 ロ 清浄区域には、排水口を設置しないこと。ただし、次に定めるところに適合する場合であつて、やむを得ないと認められるときは、この限りでない。 (1) 排水口は、清掃が容易なトラツプ及び排水の逆流を防止するための装置を有するものであること。 (2) トラツプは、消毒を行うことができる構造のものであること。 |
(続き) (3) 床の溝は、浅く清掃が容易なものであり、かつ、排水口を通じて、製造区域(培養、抽出及び精製作業、原料の秤量作業、容器の洗浄及び乾燥作業、薬剤の調製及び充填作業並びに容器の閉塞及び包装作業を行う場所並びに更衣を行う場所をいう。以下この号において同じ。)の外へ接続されていること。 ハ 無菌区域は、次に定めるところに適合するものであること。 (1) 排水口を設置しないこと。 (2) 流しを設置しないこと。 |
6構造設備 6.1 構造設備の設計上の要点 17)無菌操作区域には排水口や流しを設置しないこと。また、その他の支援区域のうちグレードCに排水口を設ける場合は清掃が容易で消毒ができるトラップ及び排水の逆流を防ぐ装置を有するなど排水口からの汚染防止を考慮すること。床に溝を設ける場合は浅く、清掃が容易な構造であること。 |
5. 建物および施設 5.1 構造設備の設計上の要点 16) グレードB以上の区域内には、流しや排水口を設けないこと。その他の支援区域(グレードC又はグレードD)に排水口を設ける場合は、清掃が容易で消毒ができるトラップ及び排水の逆流を防ぐ装置を有するなど排水口からの汚染防止を考慮すること。床に溝を設ける場合は、浅く、清掃が容易な構造であること。 |
第3章 建物及び設備 建物 製造区域 3.11. 排水溝は、適切なサイズで、トラップ付きの落とし込みを有すること。開放溝はなるべく避けるが、(必要であれば) 清浄化及び消毒を行いやすいよう浅くすること。 |
4 建物 4.9 シンク及び排水口を、グレードA及びグレードBの区域内に設置しないこと。その他のクリーンルーム内では、エアブレイクが機器又はシンクと排水口の間に取り付けられていること。低い等級のクリーンルーム内の床排水口には、逆流を防止する設計のトラップ又はウォータシールが取り付けられ、定期的に清浄化され、消毒され、保守管理されていること。 |
4.構造及び設備 4.2 ユーティリティ 4.24ドレイン配管は十分な大きさを有し、必要な場合には、逆流を防止するための空気遮断装置又は適当な装置を備えていること。 |
Subpart C - Buildings and Facilities Sec. 211.48 Plumbing. (b) Drains shall be of adequate size and, where connected directly to a sewer, shall be provided with an air break or other mechanical device to prevent back-siphonage. |
IV. BUILDINGS AND FACILITIESE. Design With rare exception, drains are considered inappropriate for classified areas of the aseptic processing facility other than Class 100,000(ISO 8) areas, It is essential that any drain installed in an aseptic processing facility be of sutitable design. |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Production Areas 3.11 Drains should be of adequate size, and have trapped gullies. Open channels should be avoided where possible, but if necessary, they should be shallow to facilitate cleaning and disinfection. |
4 Premises 4.9 Sinks and drains should be prohibited in the grade A and grade B areas. In other cleanrooms, air breaks should be fitted between the machine or sink and the drains. Floor drains in lower grade cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be regularly cleaned, disinfected and maintained. |
CHAPTER 3 PREMISES AND EQUIPMENT Production Area 3.11 Drains should be of adequate size, and have trapped gullies. Open channels should be avoided where possible, but if necessary, they should be shallow to faciliitate cleaning and disiinfection. |
4 Premises 4.9 Sinks and drains should be prohibited in the grade A and grade B areas. In other cleanrooms, air breaks should be fitted between the machine or sink and the drains. Floor drains in lower grade cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be regularly cleaned, disinfected and maintained. |
12. Premises Production areas 12.29 Drains should be of adequate size, and designed and equipped to prevent back-flow. Open channels should be avoided where possible, but if necessary, they should be shallow to faciliitate cleaning and disiinfection. |
4. Premises 4.9 Sinks and drains should be prohibited in the grade A and B areas. In other cleanrooms, air breaks should be fitted between the machine or sink and the drains. Floor drains in lower-grade cleanrooms should be fitted with traps or water seals designed to prevent backflow and should be regularly cleaned, disinfected and maintained. |
| 概 要 | 対象医薬品:原薬 以下の要件があります。 最終の精製以降の製造工程室内の排水設備について、汚染の防止が必要な構造。 *排水設備の具体的な構造は示されていません。 |
対象医薬品:特定生物由来医薬品 以下の要件があります。 ・清浄区域に、やむを得ない排水口が認められるトラツプ、逆流防止装置、床の溝についての条件について。 |
対象医薬品:特定生物由来医薬品 以下の要件があります。 ・清浄区域に、やむを得ない排水口が認められるトラツプ、逆流防止装置、床の溝についての条件について。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・グレードCおよびDに排水構造について。清掃が容易で消毒ができるトラップ、排水については、逆流を防ぐ装置、清掃がしやすい構造の溝。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・グレードCおよびDに排水構造について。清掃が容易で消毒ができるトラップ、排水については、逆流を防ぐ装置、清掃がしやすい構造の溝。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:全般 以下の要件があります。 ・適正サイズの溝かつトラップ付き。 ・開放溝は避けること(必要な場合は、清掃及び消毒がしやすい浅い構造の溝)。 |
対象医薬品:無菌医薬品 以下の要件があります。 グレードA及びグレードBを除くクリーンルーム内において流し台や排水を設置する場合、機器や流し台と排水間に、逆流防止のためのエアブレイク(逆流防止機構)を設ける。 【補足】厚生労働省通知の日本語でエアブレイクと記載されています。PIC/Sの原文はair breaksです。エアブレイク、空気遮断装置とは、逆流防止機構のことで、大気開放部の設置または機構により排水の逆流機能のあるものを示すと考えられます。 |
対象医薬品:原薬 以下の要件があります。 ・十分なサイズの排水配管。必要な場合は、空気遮断装置(逆流防止機構)を設ける。 *配管の具体的な構造は示されていません。 |
対象医薬品:全般 以下の要件があります。 ・十分なサイズの排水配管。排水管に直結されている場合は、エアブレイク(逆流防止機構)またはその他の装置を設ける。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・クラス100,000 (ISO 8) の排水は、適切な設計とすること。 *排水設備の具体的な構造は示されていません。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 製造区域 3.11と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 製造区域 3.11と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・適正サイズの溝かつ逆流防止装置付き。 ・開放溝は避けること(必要な場合は、清掃及び消毒がしやすい浅い構造の溝)。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | ICH | 日本 | 日本 | ー | ー | 欧州 | 欧州 | ー | ー | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | Q7 | 厚生労働省 | 厚生労働省 | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | ||||||||||||||||||
| 分 類 | 省令 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長通知 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||||||
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 | 薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 原薬GMPのガイドラインについて(ICHQ7) | PIC/SのGMPガイドラインを活用する際の考え方について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, No. 986 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||||||||||
| 発行年 | 2021年 | 2021年 | 2011年 | 2012年 | 2001年 | 2020年 | 2025年 | 2021年 | 2023年 | 2015年 | 2022年 | 2014年 | 2022年 | ||||||||||||||||||
| 記載内容 | 第二章 医薬品製造業者等の製造所における製造管理及び品質管理 第三節 無菌医薬品の製造管理及び品質管理 (無菌医薬品の製造所の構造設備) 第二十三条 施行規則第二十五条第一項第三号の区分の製造業者及び施行規則第三十五条第一項第三号の区分の外国製造業者の製造所の構造設備は、第九条第一項に規定するもののほか、次に定めるところに適合するものでなければならない。 四 薬剤の調製作業、充填作業、又は製品の滅菌のために行う調製作業以降の作業(表示及び包装作業を除く。)を行う作業室又は作業管理区域は、次に定めるところに適合するものであること。 イ 非無菌医薬品の作業所と区別されていること。 |
(続き) ロ 調製作業を行う作業室及び充填作業又は閉塞作業を行う作業室は専用であること。 ハ ロの作業を行う職員の専用の更衣室を有すること。 |
(続き) (生物由来医薬品等の製造所の構造設備) 第二十六条 生物由来医薬品等に係る製品の製造業者等の製造所の構造設備は、第九条第一項及び第二十三条の規定に定めるもののほか、次に定めるところに適合するものでなければならない。 二 ロットを構成しない血液製剤に係る製品の製造所の構造設備は、次に定めるところに適合するものであること。 ハ 作業所には、無菌室で作業を行う職員の専用の更衣設備を設けること。 |
第三章 医薬部外品製造業者等の製造所における製造管理及び品質管理 第三節 無菌医薬部外品の製造管理及び品質管理 (無菌医薬部外品の製造所の構造設備) 第五十一条 施行規則第二十五条第二項第一号の区分の製造業者及び施行規則第三十五条第二項第一号の区分の外国製造業者の製造所の構造設備は、第三十七条に規定するもののほか、次に定めるところに適合するものでなければならない。 四 薬剤の調製作業、充填作業、又は製品の滅菌のために行う調製作業以降の作業(表示及び包装作業を除く。)を行う作業室又は作業管理区域は、次に定めるところに適合するものであること。 イ 非無菌医薬部外品の作業所と区別されていること。 |
(続き) ロ 調製作業を行う作業室及び充填作業又は閉塞作業を行う作業室は専用であること。 ハ ロの作業を行う職員の専用の更衣室を有すること。 |
第二章 医薬品等の製造業 第一節 医薬品の製造業 (一般区分の医薬品製造業者等の製造所の構造設備) 第六条 施行規則第二十六条第一項第四号の区分及び施行規則第三十六条第一項第四号の区分の製造業者及び医薬品等外国製造業者(法第十三条の三第一項に規定する医薬品等外国製造業者をいう。)(以下「医薬品製造業者等」と総称する。)の製造所の構造設備の基準は、次のとおりとする。 三 手洗設備、便所及び更衣を行う場所を有すること。 |
6 構造設備 6.1 構造設備の設計上の要点 8) 更衣区域、衣類保管及び衣類処分のための適切な場所を設けること。 26) 更衣室はエアロックの機能を設け、脱衣と着衣のエリアを物理的に分離すること。着衣を行う部屋の微粒子清浄度はその着衣により作業する部屋の微粒子清浄度(非作業時の)と同じとすること。更衣に伴う一次的な微粒子の増加を早く低減させるため、更衣室の空間体積や換気回数(回復時間)を考慮するとともに更衣室内の空気は上部から供給し下部から排気することが望ましい。パスボックス内の清浄度は使用目的に応じて決めること。 |
(続き) 27) 特に直接支援区域の更衣室は入室と退室で分けることが望ましい。ただし、入退室の時間をずらすことで対応することもできる。 28) 更衣室は、作業する部屋の清浄度に合わせ適切に設けること。同じ清浄度内でも原材料や製品などへの交叉汚染のリスクがある場合にはリスクに応じて追加の更衣室を設けることが望ましい。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 8) 更衣区域及び作業衣保管・回収のための適切な場所を設けること。 25) 更衣室は、エアロックの機能を設け、脱衣と着衣のエリアを物理的に分離すること。着衣を行う部屋の微粒子清浄度は、その着衣により作業する部屋の微粒子清浄度(非作業時の)と同じとすることが望ましい。更衣に伴う一次的な微粒子の増加を早く低減させるため、更衣室の空間体積や換気回数(回復時間)を考慮するとともに更衣室内の空気は、上部から供給し下部から排気することが望ましい。パスボックス内の清浄度は、使用目的に応じて決めること。 |
(続き) 26) 直接支援区域をグレードBとした場合は、入室の更衣室と退室の更衣室は、分けることが望ましい。ただし、更衣室内で新しく更衣した職員と退出する職員が交差することがないように入退室の時間をずらすことで対応することもできる。 27) 更衣室は、作業する部屋の清浄度に合わせ適切に設けること。同じ清浄度内でも原材料や製品などへの交叉汚染のリスクがある場合には、リスクに応じて個別の更衣室を設けること。 |
4.構造及び設備 4.1 設計及び建設 4.15適切で清潔な手洗い設備及びトイレット設備を従業員に用意すること。これらの手洗い設備には、必要な場合には、水又は温水を備えること。また、石鹸又は洗剤並びにエアドライヤー又は使い捨てタオルを備えること。手洗い設備及びトイレット設備は作業区域から分離し、かつ、容易に利用できるように配置すること。必要な場合は、シャワーや更衣のための適切な設備を設置すること。 |
第3章 建物及び設備 建物 付随区域 3.31. 更衣施設並びに手洗い及びトイレの施設は、容易にアクセス可能であり、使用者数に対し適切な数があること。トイレは、製造及び貯蔵区域と直接通じていてはならない。 |
4 建物 4.12 (略) グレードB区域を入退室する際には、別々の更衣室を使うことが望ましい。それが実践的でない場合には、(入室/退室)作業の時間を別にする手順を検討すること。CCSで汚染のリスクが高いことが示されている場合には、製造区域の入退室で別々の更衣室が使われること。エアロックは、以下のように設計されていること: i.人員用のエアロック: 人員の立入りに使われる、清浄度が次第に高くなる区域(例:グレードD区域からグレードC区域へ、続いてグレードB区域へ)。一般に手洗い施設は、更衣室の第一段階にのみ設けることとし、グレードB区域に直接つながっている更衣室内にあってはならない。 |
7 人員 7.10 クリーンルームの作業衣着用及び手洗いは、クリーンルーム着衣の汚染及び/又は汚染物質の清浄区域への移行を最小化するように設計された手順書に従うこと。 |
(続き) 7.14 クリーンルームの作業衣着用は適切な清浄度等級の更衣室内で行って、作業衣の清浄度が保持されていることが確保されていること。靴下等の室外着衣(個人の下着を除く)がグレードB及びCの区域に直接つながる更衣室内に持ち込まれてはならない。上下つなぎ又はツーピースの施設ズボンスーツ(腕部及び脚部の全体を覆うもの)及び足部を覆う施設靴下を、グレードB及びCのための更衣室へ入る前に着用すること。施設スーツ及び施設靴下が、作業衣着用の区域又は過程に汚染のリスクをもたらすものであってはならない。 |
用語解説 汚染制御ストラテジー(CCS) - 微生物、エンドトキシン/発熱性物質及び微粒子を制御する計画一式であり、現行の製品及び工程の理解から導き出され、工程性能及び製品品質を保証するもの。当該制御には、原薬、添加剤並びに薬剤の原材料及び構成物、施設及び設備の操作条件、工程内管理、最終製品の規格、並びに付随するモニタリング及び管理の頻度及び方法に関連するパラメータ及び特性が含まれ得る。 |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Ancillary Areas 3.31. Facilities for changing clothes and for washing and toilet purposes should be easily accessible and appropriate for the number of users. Toilets should not directly communicate with production or storage areas. |
4 Premises 4.12 (略) The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (ingress/egress) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. Airlocks should be designed as follows: i. Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. |
7 Personnel 7.10 Cleanroom gowning and hand washing should follow a written procedure designed to minimize contamination of cleanroom clothing and/or the transfer of contaminants to the clean areas. |
(続き) 7.14 Cleanroom gowning should be performed in change rooms of an appropriate cleanliness grade to ensure gown cleanliness is maintained. Outdoor clothing including socks (other than personal underwear) should not be brought into changing rooms leading directly to grade B and C areas. Single or two-piece facility trouser suits, covering the full length of the arms and the legs, and facility socks covering the feet, should be worn before entry to change rooms for grades B and C. Facility suits and socks should not present a risk of contamination to the gowning area or processes. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
CHAPTER 3 PREMISES AND EQUIPMENT Premises Ancillary areas 3.31 Facilities for changing clothes and for washing and toilet purposes should be easily accessible and appropriate for the number of users. Toilets should not directly communicate with production or storage areas. |
4 Premises 4.12 (略) The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (ingress/egress) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. Airlocks should be designed as follows: i. Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. |
7 Personnel 7.10 Cleanroom gowning and hand washing should follow a written procedure designed to minimize contamination of cleanroom clothing and/or the transfer of contaminants to the clean areas. |
(続き) 7.14 Cleanroom gowning should be performed in change rooms of an appropriate cleanliness grade to ensure gown cleanliness is maintained. Outdoor clothing including socks (other than personal underwear) should not be brought into changing rooms leading directly to grade B and C areas. Single or two-piece facility trouser suits, covering the full length of the arms and the legs, and facility socks covering the feet, should be worn before entry to change rooms for grades B and C. Facility suits and socks should not present a risk of contamination to the gowning area or processes. |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
12.Premises Ancillary areas 12.12 Facilities for changing and storing clothes and for washing and toilet purposes should be easily accessible and appropriate for the number of users. Toilets should not communicate directly with production or storage areas. |
4. Premises 4.12 (略) The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (inward or outward) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. Airlocks should be designed as follows: i. Personnel airlocks: areas of increasing cleanliness used for entry of personnel (for example, from the grade D area to the grade C area to the grade B area). In general, handwashing facilities should be provided only in the first change room and should not be present in change rooms directly accessing the grade B area. |
7. Personnel 7.10 Cleanroom gowning and handwashing should follow a written procedure designed to minimize contamination of cleanroom clothing or the transfer of contaminants to the clean areas. |
(続き) 7.14 Cleanroom gowning should be performed in change rooms of an appropriate cleanliness grade to ensure that gown cleanliness is maintained. Outdoor clothing, including socks (other than personal underwear), should not be brought into changing rooms leading directly to grade B and C areas. Single- or two-piece facility trouser suits, covering the full length of the arms and the legs, and facility socks covering the feet should be worn before entry to change rooms for grades B and C. Facility suits and socks should not present a risk of contamination to the gowning area or processes. |
Glossary contamination control strategy (CCS). A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
| 概 要 | ー | 対象医薬品:無菌医薬品 以下の要件があります。 ・調製作業及び充填作業又は閉塞作業を行う作業員の専用更衣室を設ける。 |
対象医薬品:生物由来医薬品等 以下の要件があります。 ・無菌室で作業を行う職員の専用の更衣設備を設ける。 |
ー | 対象医薬品:無菌医薬部外品 以下の要件があります。 ・調製作業及び充填作業又は閉塞作業を行う作業員の専用更衣室を設ける。 |
対象医薬品:全般 以下の要件があります。 手洗設備、更衣室設備について、必要とされています。 *具体的な構造は示されていません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・脱衣と着衣のエリアを物理的に分離すること。 ・更衣室はエアロックの機能があること。 ・交叉汚染のリスクがある場合にはリスクに応じて追加の更衣室すること。 ・更衣室内の空気は上部から供給し下部から排気すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・直接支援区域の更衣室は入室と退室で分けることが望ましい。分離しない更衣室は、入退室の時間差の対応で可能。 ・清浄度はその着衣により作業する部屋の微粒子清浄度(非作業時の)と同じとすること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・脱衣と着衣のエリアを物理的に分離すること。 ・更衣室はエアロックの機能があること。 ・交叉汚染のリスクがある場合にはリスクに応じて追加の更衣室すること。 ・更衣室内の空気は上部から供給し下部から排気すること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・直接支援区域をグレードBとした場合の更衣室は入室と退室で分けることが望ましい。分離しない更衣室は、入退室の時間差の対応で可能。 ・清浄度はその着衣により作業する部屋の微粒子清浄度(非作業時の)と同じとすること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:原薬 以下の要件があります。 ・必要な場合は、シャワーや更衣のための適切な設備を設置すること。 |
対象医薬品:全般 以下の配置要件があります。 ・更衣室は容易にアクセスできること。 ・適切な数があること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードBの更衣室は入室と退室で分けることが望ましい。CCSに基づき、分離を対応する。分離が実践的でない更衣室は、入退室の時間差の手順を検討すること。 ・手洗い設備は更衣室の初めの段階のみとして、グレードBに直接アクセスする更衣室には設置しないこと。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームのための更衣と手洗いは、クリーン更衣やクリーンエリアの汚染を最小限に抑える設計された手順とすること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの更衣は、更衣の清潔さを保証するため、適切なグレードの更衣室で行うこと。 ・靴下を含む外着(個人用下着以外)は、グレードBおよびCエリアに直接つながる更衣室に持ち込まないようにすること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCSの定義。 |
対象医薬品:全般 別紙(1)PIC/S GMP ガイドライン パート 1 第3章 建物及び設備 建物 付随区域 3.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:全般 別紙(1)PIC/S GMP ガイドライン パート 1 第3章 建物及び設備 建物 付随区域 3.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・更衣室は容易にアクセスできること。 ・使用人数に対して、適切な数があること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.10と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | 日本 | ー | ー | 欧州 | 欧州 | ー | ー |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | 厚生労働省 | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO |
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Code of Federal Regulations | 厚生労働省医薬局長通知 | GMP | GMP | GMP | GMP | GMP | GMP |
| 法規類 タイトル |
薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
原薬GMPのガイドラインについて(ICHQ7) | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, No. 986 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
| 発行年 | 2021年 | 2011年 | 2012年 | 2020年 | 2025年 | 2023年 | 2001年 | 2021年 | 2023年 | 2015年 | 2022年 | 2014年 | 2022年 |
| 記載内容 | 第二章 医薬品等の製造業 第一節 医薬品の製造業 (一般区分の医薬品製造業者等の製造所の構造設備) 第六条 施行規則第二十六条第一項第四号の区分及び施行規則第三十六条第一項第四号の区分の製造業者及び医薬品等外国製造業者(法第十三条の三第一項に規定する医薬品等外国製造業者をいう。)(以下「医薬品製造業者等」と総称する。)の製造所の構造設備の基準は、次のとおりとする。 三 手洗設備、便所及び更衣を行う場所を有すること。 |
6 構造設備 6.1 構造設備の設計上の要点 1) 清浄区域はトイレ、飲食等を行う場所から明確に区分されていること。 |
5 構造設備 5.1 構造設備の設計上の要点 1) 清浄区域は、トイレ、飲食等を行う場所から明確に区分されていること。 |
第3章 建物及び設備 建物 付随区域 3.31. 更衣施設並びに手洗い及びトイレの施設は、容易にアクセス可能であり、使用者数に対し適切な数があること。トイレは、製造及び貯蔵区域と直接通じていてはならない。 |
4 建物 4.12 (略) i 人員用のエアロック: 人員の立入りに使われる、清浄度が次第に高くなる区域(例:グレードD区域からグレードC区域へ、続いてグレードB区域へ)。一般に手洗い施設は、更衣室の第一段階にのみ設けることとし、グレードB区域に直接つながっている更衣室内にあってはならない。 7 人員 7.10 クリーンルームの作業衣着用及び手洗いは、クリーンルーム着衣の汚染及び/又は汚染物質の清浄区域への移行を最小化するように設計された手順書に従うこと。 |
Subpart C - Buildings and Facilities Sec. 211.52 Washing and toilet facilities. Adequate washing facilities shall be provided, including hot and cold water, soap or detergent, air driers or single-service towels, and clean toilet facilities easily accesible to working areas. |
4.構造及び設備 4.1 設計及び建設 4.15適切で清潔な手洗い設備及びトイレット設備を従業員に用意すること。これらの手洗い設備には、必要な場合には、水又は温水を備えること。また、石鹸又は洗剤並びにエアドライヤー又は使い捨てタオルを備えること。手洗い設備及びトイレット設備は作業区域から分離し、かつ、容易に利用できるように配置すること。必要な場合は、シャワーや更衣のための適切な設備を設置すること。 |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Ancillary Areas 3.31Facilities for changing clothes and for washing and toilet purposes should be easily accessible and appropriate for the number of users. Toilets should not directly communicate with production or storage areas. |
4 Premises 4.12 (略) i. Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. 7 Personnel 7.10 Cleanroom gowning and hand washing should follow a written procedure designed to minimize contamination of cleanroom clothing and/or the transfer of contaminants to the clean areas. |
CHAPTER 3 PREMISES AND EQUIPMENT Premises Ancillary Areas 3.31 Facilities for changing clothes and for washing and toilet purposes should be easily accessible and appropriate for the number of users. Toilets should not directly communicate with production or storage areas. |
4 Premises 4.12 (略) i. In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. 7 Personnel 7.10 Cleanroom gowning and hand washing should follow a written procedure designed to minimize contamination of cleanroom clothing and/or the transfer of contaminants to the clean areas. |
12.Premises Ancillary areas 12.12 Facilities for changing and storing clothes and for washing and toilet purposes should be easily accessible and appropriate for the number of users. Toilets should not communicate directly with production or storage areas. |
4. Premises 4.12 (略) i. In general, handwashing facilities should be provided only in the first change room and should not be present in change rooms directly accessing the grade B area. 7. Personnel 7.10 Cleanroom gowning and handwashing should follow a written procedure designed to minimize contamination of cleanroom clothing or the transfer of contaminants to the clean areas. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・手洗設備、トイレが必要。 *「手洗設備、更衣室」の具体的な構造、構成の記載はありません。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・トイレは、清浄区域と明確に区分すること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・トイレは、清浄区域と明確に区分すること。 |
対象医薬品:全般 以下の要件があります。 ・トイレ、手洗設備は、容易にアクセスできること。 ・トイレ、手洗設備は、 使用人数に対して、適切な数があること。 ・トイレは、製造又は貯蔵区域と直接通じていないこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードBに直接アクセスする更衣室には手洗い設備は、設置不可。更衣室構成の第一段階にのみ設置すること。 ・作業衣の汚染防止と汚染物質の持込防止を考慮した手洗いの手順について。 |
対象医薬品:全般 以下の要件があります。 ・手洗設備、トイレが必要。 ・手洗設備は、温水と水、石鹸又は洗剤並びにエアドライヤー又は使い捨てタオルの設備があること。 ・清潔なトイレが作業区域から容易に利用できること。 |
対象医薬品:原薬 以下の要件があります。 ・手洗設備、トイレが必要。 手洗設備として、必要な場合は ・水または温水があること。 ・石鹸または洗剤、エアドライヤーまたは使い捨てタオルの設備があること。 ・手洗設備及びトイレは作業区域から分離し、かつ、容易に利用できること。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 7 人員 7.10と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 7 人員 7.10と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.31と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 7 人員 7.10と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | ー | 欧州 | ー |
|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO |
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP |
| 法規類 タイトル |
薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, No. 986 2014 |
| 発行年 | 2021年 | 2011年 | 2012年 | 2020年 | 2021年 | 2015年 | 2014年 |
| 記載内容 | 第二章 医薬品等の製造業 第一節 医薬品の製造業 (一般区分の医薬品製造業者等の製造所の構造設備) 第六条 施行規則第二十五条第一項第四号の区分及び施行規則第三十五条第一項第四号の区分の製造業者及び医薬品等外国製造業者(法第十三条の三第一項に規定する医薬品等外国製造業者をいう。)(以下「医薬品製造業者等」と総称する。)の製造所の構造設備の基準は、次のとおりとする。 四 製造作業を行う場所(以下「作業所」という。)は、次に定めるところに適合するものであること。 ロ 常時居住する場所及び不潔な場所から明確に区別されていること。 |
6 構造設備 6.1 構造設備の設計上の要点 1) 清浄区域はトイレ、飲食等を行う場所から明確に区分されていること。 |
5. 構造設備 5.1 構造設備の設計上の要点 1) 清浄区域は、トイレ、飲食等を行う場所から明確に区分されていること。 |
第3章 建物及び設備 建物 付随区域 3.30. 休憩室は、他の区域と別にすること。 |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES Ancillary Areas 3.30Rest and refreshment rooms should be separate from other areas. |
CHAPTER 3 PREMISES AND EQUIPMENT Premises Ancillary Areas 3.30 Rest and refreshment rooms should be separate from other areas. |
12.Premises Ancillary areas 12.11 Rest and refreshment rooms should be separate from manufacturing and control areas. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・「休憩室」と明記されていませんが、常時居住する場所は、製造作業を行う場所と明確に区別されていること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・「休憩室」と明記されていませんが、飲食等を行う場所は、清浄区域と明確に区分されていること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・「休憩室」と明記されていませんが、飲食等を行う場所は、清浄区域と明確に区分されていること。 |
対象医薬品:全般 以下の要件があります。 ・休憩室は、他の区域と分離されていること。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.30と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.30と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・休憩/リフレッシュルームは、製造、管理の区域と分離されていること。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | ー | 欧州 | ー | ー | ー | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO | WHO | WHO | ||
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | GMP | ||
| 法規類 タイトル |
薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(3)-2 PIC/S GMPガイドライン アネックス2B |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||
| 発行年 | 2021年 | 2011年 | 2012年 | 2025年 | 2022年 | 2023年 | 2022年 | 2018年 | 2019年 | 2022年 | ||
| 記載内容 | 第二章 医薬品等の製造業 第一節 医薬品の製造業 (一般区分の医薬品製造業者等の製造所の構造設備) 第六条 施行規則第二十五条第一項第四号の区分及び施行規則第三十五条第一項第四号の区分の製造業者及び医薬品等外国製造業者(法第十三条の三第一項に規定する医薬品等外国製造業者をいう。)(以下「医薬品製造業者等」と総称する。)の製造所の構造設備の基準は、次のとおりとする。 5 原薬に係る製品の作業所のうち、最終の精製以後の製造工程において、最終の精製を経た中間製品を容器へ充填及び閉塞するまでの作業を行う作業室及び原薬に係る製品以外の製品の作業所のうち、原料の秤量作業、製品の調製作業、充填作業又は閉塞作業を行う作業室は、次に定めるところに適合するものであること。 |
イ 屋外に直接面する出入口(非常口を除く。)がないこと。ただし、屋外からの汚染を防止するのに必要な構造及び設備を有している場合においては、この限りでない。 | (放射性医薬品区分の医薬品製造業者等の製造所の構造設備) 第九条 施行規則第二十五条第一項第二号の区分及び施行規則第三十五条第一項第二号の区分の医薬品製造業者等の製造所(包装、表示又は保管のみを行う製造所を除く。以下この項及び次項において同じ。)の構造設備の基準は、第六条及び第七条に定めるもののほか、次のとおりとする。 三 次に定めるところに適合する貯蔵設備を有すること。 ニ 扉、蓋等外部に通ずる部分に、鍵その他閉鎖のための設備又は器具を有すること。 |
6 構造設備 6.1 構造設備の設計上の要点 7) 粒子あるいは微生物がたまったり気流を妨げたりする可能性のある凹凸構造、窓、扉周り等の横桟の設置は可能な限り避けること。やむを得ない場合は容易に清掃できる構造とすること。特に無菌操作区域にはスライディングドアは好ましくない。 |
5 建物及び施設 5.1 構造設備の設計上の要点 7) 粒子あるいは微生物がたまったり気流を妨げたりする可能性のある凹凸構造、窓、扉周り等の横桟の設置は、可能な限り避けること。やむを得ず設置する場合は、容易に清掃できる構造とすること。 |
4 建物 4.6 塵埃が溜まるのを低減し、清浄化し易くするため、効果的に清浄化することが難しい凹みがないこと。よって、突き出た棚、棚板、戸棚及び設備は最小限度にしておくこと。ドアは、清浄化できない凹みがないように設計されていること。そのため、スライドドアは望ましくないことがあり得る。 |
パートA. 一般的ガイダンス 作業原則 50. 製造区域内への物品及び原材料の搬入に関する管理ストラテジーは、QRMの原則に基づくものであること。無菌工程については、耐熱性のある物品及び原材料を清浄区域又は清浄/封じ込め区域に搬入する際に、取入れ口と取出し口が別になっている(double-ended)オートクレーブ又はオーブンにかけることが望ましい。耐熱性のない物品及び原材料は、インターロックドア付きのエアロックを通して搬入することとし、そのドアは、所定の手順で表面を効果的に殺菌すること。別の場所で物品及び原材料を滅菌することは許容され得るが、清浄区域への搬入段階の数に応じて、当該物品及び原材料が幾重にも包装されており、適切な表面消毒の予防策を講じた上でエアロックを通して搬入すること。 |
4 Premises 4.6 To reduce accumulation of dust and to facilitate cleaning there should be no recesses that are difficult to clean effectively, therefore projecting ledges, shelves, cupboards and equipment should be kept to a minimum. Doors should be designed to avoid recesses that cannot be cleaned. Sliding doors may be undesirable for this reason. |
4 Premises 4.6 To reduce accumulation of dust and to facilitate cleaning there should be no recesses that are difficult to clean effectively, therefore projecting ledges, shelves, cupboards and equipment should be kept to a minimum. Doors should be designed to avoid recesses that cannot be cleaned. Sliding doors may be undesirable for this reason. |
4. Premises 4.13 Where appropriate, such as where the simultaneous opening of airlock doors might lead to a cross-contamination risk, airlock doors should not be opened at the same time. In such cases, controls such as interlocking systems, warning systems and procedures should be implemented. 4.14 Swing doors should normally open to the high-pressure side and be provided with self-closers. Exceptions to the door swing direction should be justified and may include for fire escapes or other health and safety measures. In these cases, door closer mechanisms should be carefully controlled and other controls should be in place to prevent any risk. |
4. Premises 4.1 Premises design (省略) Where necessary, air locks, change rooms and pass-through hatches may be considered and provided with effective ventilation and filtered air. Special attention should be given to door design, as gaps between doors and floors, doors opening into low-pressure areas, and sliding doors can result in changes in the pressure differential between areas. An interlocking system and a visual and/or audible warning system may be used, where required, to prevent opening of more than one door at a time where required. |
4. Premises 4.6 To reduce accumulation of dust and to facilitate cleaning, there should be no recesses that are difficult to clean effectively. Projecting ledges, shelves, cupboards and equipment should be kept to a minimum. Doors should be designed to avoid recesses that cannot be cleaned. Sliding doors may be undesirable for this reason. |
| 概 要 | ー | 対象医薬品:原薬 以下の要件があります。 ・屋外に直接面する出入口がないこと。(ただし非常口については適用外) |
対象医薬品:放射性医薬品 以下の要件があります。 ・扉等外部に通ずる部分には、鍵その他の閉鎖のための設備又は器具の設けられていること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・扉周り等の横桟の設置は可能な限り避けること。やむを得ない場合は容易に清掃できる構造とすること。 ・無菌操作区域には引き戸は好ましくない。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・扉周り等の横桟の設置は可能な限り避けること。やむを得ない場合は容易に清掃できる構造とすること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・扉は清浄化できない凹部みはないように設計すること。 ・引き戸は好ましくはない。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 以下の要件があります。 ・無菌工程における製造区域への物品及び原材料の搬入は、インターロックドア付きのエアロックを通じて搬入すること。 ・エアロックの扉は、所定の手順で表面を効果的に殺菌すること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.6と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.6と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・エアロックの扉の同時開放を防止する インターロックシステムや警報システムを使用すること。 ・扉は、高圧側に向かう開き方向とすること。 ・扉は、セルフクローザー付(自動閉鎖装置)とすること。 *用語についての補足:「エアロック」は 建築 007:エアロックのWHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・扉と床の隙間、低圧側に向かう開き方向、引き戸は、区域間の差圧に影響するなど、設計の留意事項にについて。 ・エアロック扉の同時開放を防止する インターロックシステムや視覚的/聴覚的警報システムを使用すること。 *用語についての補足:「エアロック」は 建築 007:エアロックのWHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.6と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | ー | ー | ー | 欧州 | 欧州 | ー | ー | ー | ー | ー | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | CDER | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | WHO | WHO | WHO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(3)-2 PIC/S GMPガイドライン アネックス2B |
「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(9) PIC/S GMPガイドライン アネックス10 定量噴霧式吸入剤の製造 |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 2B MANUFACTURE OF BIOLOGICAL MEDICINAL SUBSTANCES AND PRODUCTS FOR HUMAN USE |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 10 MANUFACTURE OF PRESSURISED METERED DOSE AEROSOL PREPARATIONS FOR INHALATION |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 10 MANUFACTURE OF PRESSURISED METERED DOSE AEROSOL PREPARATIONS FOR INHALATION |
WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS 48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, 986 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
44th report Annex 3 WHO good manufacturing practices for pharmaceutical products containing hazardous substances WHO Technical Report Series, 957 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2011年 | 2012年 | 2025年 | 2022年 | 2012年 | 2004年 | 2023年 | 2023年 | 2023年 | 2023年 | 2009年 | 2014年 | 2010年 | 2018年 | 2019年 | 2022年 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 記載内容載内容 |
6. 構造設備 6.1 構造設備の設計上の要点 24) 直接支援区域と直接支援区域に隣接する区域とはエアロックにより分離すること。直接支援区域と直接支援区域に隣接する部屋との間には、滅菌済み資材、滅菌が困難な資材等の受渡し及び必要な場合においては除染作業等のためのパスルームやパスボックスを設けること。 25) エアロック扉には同時に開かないような装置(機械式、電気式のほか目視又は音を利用した方法等)を備えること。 |
26) 更衣室はエアロックの機能を設け、脱衣と着衣のエリアを物理的に分離すること。 着衣を行う部屋の微粒子清浄度はその着衣により作業する部屋の微粒子清浄度(非作業時の)と同じとすること。更衣に伴う一次的な微粒子の増加を早く低減させるため、更衣室の空間体積や換気回数(回復時間)を考慮するとともに更衣室内の空気は上部から供給し下部から排気することが望ましい。パスボックス内の清浄度は使用目的に応じて決めること。 | 7. 無菌医薬品に係る製品の作業所 7.2 空調システム 7.2.2 空気 2) 無菌操作区域とその他の支援区域との間にはエアロックを設け、室間差圧及び気流の逆転が起きないよう、十分な差圧を設けること。扉を閉じた状態で10〜15Pa又はそれ以上の差圧を維持することが望ましい。当該エアロックの設計は、6.1 26)更衣室に準じる。 その他の支援区域内においても空気の清浄度レベルの異なる区域の間には、適切な差圧を設けること。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 23) 直接支援区域と直接支援区域に隣接するその他の区域の清浄度が異なる場合は、エアロックにより分離すること。同じ清浄度の場合は、それぞれの区域での作業内容による影響を勘案しエアロックを設けること。直接支援区域と直接支援区域に隣接する部屋との間において、滅菌済み資材の受渡しを行う場合は、パスルームやパスボックスを設けること。滅菌済み資材や滅菌が困難な資材(測定機器類等を含む)の受渡しで外装の除染が必要な場合においては、パスルームやパスボックス内で除染作業等も行えるよう考慮すること。 24) エアロック扉には、同時に開かないような装置(機械式、電気式等)のほか目視又は音を利用した方法等を備えること。 |
25) 更衣室は、エアロックの機能を設け、脱衣と着衣のエリアを物理的に分離すること。着衣を行う部屋の微粒子清浄度は、その着衣により作業する部屋の微粒子清浄度(非作業時の)と同じとすることが望ましい。更衣に伴う一次的な微粒子の増加を早く低減させるため、更衣室の空間体積や換気回数(回復時間)を考慮するとともに更衣室内の空気は、上部から供給し下部から排気することが望ましい。パスボックス内の清浄度は、使用目的に応じて決めること。 | 4 建物 4.1 無菌製品の製造が適切なクリーンルーム内で行われていること、当該クリーンルームには、エアロックとして機能する更衣室を通って人員が入室し、且つ設備及び原材料がエアロックを通して搬入されていること。クリーンルーム及び更衣室は、適切な清浄度基準に維持管理され、且つ適切な効率のフィルタを通った空気が供給されていること。管理及び監視では、科学的に妥当性を示すこと、且つクリーンルーム、エアロック及びパススルーハッチについての環境条件の状態を効果的に評価すること。 |
4.12 エアロックは、物理的な分離を与えるように、且つ、異なる区域の微生物・微粒子汚染が最小化されるように設計され、使用されていること、且つ異なる清浄度等級間を移動する原材料及び人員に供されていること。人員の移動用のエアロックは、なるべく原材料の移動用のエアロックと別になっていること。それが実践的でない場合には、(人員/原材料の)移動する時間を別にする手順を検討すること。 | (続き) エアロックをフィルタ処理された空気で効果的に換気して、クリーンルームの等級が維持管理されていることを確保にすること。エアロックの最終段階は、「非作業時」の状態において、そこからつながるクリーンルームと同じ清浄度等級(生菌数及び総微粒子量)になっていること。グレードB区域を入退室する際には、別々の更衣室を使うことが望ましい。それが実践的でない場合には、(入室/退室) 作業の時間を別にする手順を検討すること。CCSで汚染のリスクが高いことが示されている場合には、製造区域の入退室で別々の更衣室が使われること。 |
(続き) エアロックは、以下のように設計されていること: i.人員用のエアロック: 人員の立入りに使われる、清浄度が次第に高くなる区域(例:グレードD区域からグレードC区域へ、続いてグレードB区域へ)。一般に手洗い施設は、更衣室の第一段階にのみ設けることとし、グレードB区域に直接つながっている更衣室内にあってはならない。 ii.原材料用のエアロック: 原材料及び設備の搬送に使われる。 ・承認を受けたリストに収載されている原材料及び設備であって搬送工程のバリデーションの際に評価済みであるもののみが、グレードA又はグレードBの区域内へエアロック又はパススルーハッチを通して搬入されていること。 |
(続き) 設備及び原材料(グレードA区域内で用いようとするもの) がグレードB区域を通過する際に保護されていること。承認を受けていない物品で搬送を要するものがあれば、例外として事前承認を受けること。 リスクの適切な評価及び軽減措置は、その製造業者のCCSに則って適用され且つ記録作成されていること、また、品質保証担当によって承認を受けた特別な消毒及びモニタリングのプログラムを含めること。 |
(続き) ・パススルーハッチは、(例えば、有効なフィルタ処理された給気で効果的に換気することで)より高い等級の環境を保護するように設計されていること。 ・より低い等級の区域又は等級分けされていない区域からより高い等級の区域への原材料又は設備の搬送は、CCSに沿ってリスクに相応した清浄化及び消毒の対象となっていること。 |
4.13 パススルーハッチ及び(原材料用及び人員用の)エアロックについて、入口及び出口のドアを同時に開けてはならない。グレードA及びグレードBの区域につながるエアロックには、インターロックのシステムが用いられること。グレードC及びDの区域につながるエアロックについては最低限、視覚的及び/又は聴覚的な警報システムを作動させること。区域の隔離を保持することが求められる場合には、インターロック付きドアの開閉する間で時間の遅延が確立していること。 | 4.14 クリーンルームは、全ての作業条件下で、より低い等級のバックグラウンド環境に比して陽圧及び/又は気流を保持するフィルタ処理された給気が供給されていること、また、その給気は当該区域を効果的に換気するものであること。異なる清浄度等級の隣接する部屋には、少なくとも10パスカル(ガイダンス値) の気圧差があること。重要区域の保護には、特別な注意を払うこと。ある種の原材料(例: 病原性、高毒性若しくは放射性の生成物、又は生きたウイルス若しくは細菌性の原材料) を封じ込めることが必要な場合において、給気及び気圧に関する推奨事項を修正しなければならないことがあり得る。当該修正には、危険物質が周囲の区域を汚染するのを防止する陽圧又は陰圧がかかったエアロックが含まれうる。 | (続き) 施設(例:クリーンルーム及び暖房、換気、空調(HVAC)システム)の除染及び清浄区域から出ていく空気の処理が必要とされ得る作業もある。封じ込めで重要区域に空気を流入させることを要する場合には、当該空気の供給源は、同等以上の等級の区域からとすること。 |
用語解説 エアロック - インターロック付きドアのある囲まれた空間で、隣接する室(一般に、異なる空気清浄基準が異なるもの)間の気圧制御を保持するよう構築されたもの。エアロックの目的は、より低い管理がなされる区域から微粒子物質及び微生物の汚染が入り込むのを防ぐことである。 |
用語解説 パススルーハッチ - エアロック(エアロックの定義を参照) と同義であるが、一般的に寸法が小さいもの。 |
パートA. 一般的ガイダンス 作業原則 50. 製造区域内への物品及び原材料の搬入に関する管理ストラテジーは、QRMの原則に基づくものであること。無菌工程については、耐熱性のある物品及び原材料を清浄区域又は清浄/封じ込め区域に搬入する際に、取入れ口と取出し口が別になっている(double-ended)オートクレーブ又はオーブンにかけることが望ましい。耐熱性のない物品及び原材料は、インターロックドア付きのエアロックを通して搬入することとし、そのドアは、所定の手順で表面を効果的に殺菌すること。別の場所で物品及び原材料を滅菌することは許容され得るが、清浄区域への搬入段階の数に応じて、当該物品及び原材料が幾重にも包装されており、適切な表面消毒の予防策を講じた上でエアロックを通して搬入すること。 |
建物及び設備 3. 製品又は洗浄済みの構成部品が曝される区域にはろ過された空気を供給し、その区域は、少なくともグレードD環境の要求を満たし、エアロックを介して入室しなければならない。 |
IV. BUILDINGS AND FACILITIES E. Design (略) Use of a double-door or integrated sterilizer helps ensure direct product flow, often from a lower to a higher classified area. Airlocks and interlocking doors will facilitate better control of air balance throughout the aseptic processing facility. Airlocks should be installed between the aseptic manufacturing area entrance and the adjoining unclassified area. Other interfaces such as personnel transitions or material staging areas are appropriate locations for air locks. |
GLOSSARY Air lock - A small room with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an aseptic processing airlock is to preclude ingress of particulate matter and microorganism contamination from a lesser controlled area. |
4 Premises 4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to which should be through change rooms that act as airlocks for personnel and airlocks for equipment and materials. Cleanrooms and change rooms should be maintained to an appropriate cleanliness standard and supplied with air which has passed through filters of an appropriate efficiency. Controls and monitoring should be scientifically justified and should effectively evaluate the state of environmental conditions of cleanrooms, airlocks and pass-through hatches. |
4.12 Airlocks should be designed and used to provide physical separation and to minimize microbial and particle contamination of the different areas and should be present for material and personnel moving between different grades. Wherever possible, airlocks used for personnel movement should be separated from those used for material movement. Where this is not practical, time-based separation of movement (personnel/material) by procedure should be considered. | (続き) Airlocks should be flushed effectively with filtered air to ensure that the grade of the cleanroom is maintained. The final stage of the airlock should, in the “at rest” state, be of the same cleanliness grade (viable and total particle) as the cleanroom into which it leads. The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (ingress/egress) by procedure should be considered.Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. |
(続き) Airlocks should be designed as follows: i.Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. ii.Material airlocks: used for materials and equipment transfer. ・Only materials and equipment that have been included on an approved list and assessed during validation of the transfer process, should be transferred into the grade A or grade B areas via an airlock or pass-through hatches. |
(続き) Equipment and materials (intended for use in the grade A area) should be protected when transiting through the grade B area. Any unapproved items that require transfer should be pre-approved as an exception. Appropriate risk assessment and mitigation measures should be applied and recorded as per the manufacturer's CCS and should include a specific disinfection and monitoring programme approved by quality assurance. |
(続き) ・Pass-through hatches should be designed to protect the higher-grade environment, for example by effective flushing with an active filtered air supply. ・The movement of material or equipment from lower grade or unclassified area to higher grade clean areas should be subject to cleaning and disinfection commensurate with the risk and in line with the CCS. |
4.13 For pass-through hatches and airlocks (for material and personnel), the entry and exit doors should not be opened simultaneously. For airlocks leading to the grade A and grade B areas, an interlocking system should be used. For airlocks leading to grade C and D areas, a visual and/or audible warning system should be operated as a minimum. Where required to maintain area segregation, a time delay between the closing and opening of interlocked doors should be established. | 4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of different grades should have an air pressure difference of a minimum of 10 Pascals (guidance value). Particular attention should be paid to the protection of the critical zone. The recommendations regarding air supplies and pressures may need to be modified where it is necessary to contain certain materials (e.g. pathogenic, highly toxic or radioactive products or live viral or bacterial materials). The modification may include positively or negatively pressurized airlocks that prevent the hazardous material from contaminating surrounding areas. | (続き) Decontamination of facilities (e.g. the cleanrooms and the heating, ventilation, and air conditioning (HVAC) systems) and the treatment of air leaving a clean area, may be necessary for some operations. Where containment requires air to flow into a critical zone, the source of the air should be from an area of the same or higher grade. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
PART A: GENERAL GUIDANCE OPERATING PRINCIPLES 50.A control strategy for the entry of articles and materials into production areas should be based on QRM principles. For aseptic processes, heat stable articles and materials entering a clean area or clean/contained area should preferably do so through a double-ended autoclave or oven. Heat labile articles and materials should enter through an air lock with interlocked doors where they are subject to effective surface sanitisation procedures. Sterilisation of articles and materials elsewhere is acceptable provided that they are multiple wrappings, as appropriate to the number of stages of entry to the clean area, and enter through an airlock with the appropriate surface sanitisation precautions. |
PREMISES AND EQUIPMENT 3.Where products or clean components are exposed, the area should be fed with filtered air, should comply with the requirements of at least a Grade D environment and should be entered through airlocks. |
4 PremisesT 4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to which should be through change rooms that act as airlocks for personnel and airlocks for equipment and materials. Cleanrooms and change rooms should be maintained to an appropriate cleanliness standard and supplied with air that has passed through filters of an appropriate efficiency. Controls and monitoring should be scientifically justified and should effectively evaluate the state of environmental conditions of cleanrooms, airlocks and pass-through hatches. |
4.12 Airlocks should be designed and used to provide physical separation and to minimize microbial and particle contamination of the different areas and should be present for material and personnel moving between different grades. Wherever possible, airlocks used for personnel movement should be separated from those used for material movement. Where this is not practical, time-based separation of movement (personnel/material) by procedure should be considered. | (続き) Airlocks should be flushed effectively with filtered air to ensure that the grade of the cleanroom is maintained. The final stage of the airlock should, in the “at rest” state, be of the same cleanliness grade (viable and total particle) as the cleanroom into which it leads. The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (ingress/egress) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. |
(続き) Airlocks should be designed as follows: i.Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from the grade D area to the grade C area to the grade B area). In general hand washing facilities should be provided only in the first stage of the changing room and not be present in changing rooms directly accessing the grade B area. ii.Material airlocks: used for materials and equipment transfer. ・Only materials and equipment that have been included on an approved list and assessed during validation of the transfer process should be transferred into the grade A or grade B areas via an airlock or pass-through hatches. |
(続き) Equipment and materials (intended for use in the grade A area) should be protected when transiting through the grade B area. Any unapproved items that require transfer should be pre-approved as an exception. Appropriate risk assessment and mitigation measures should be applied and recorded as per the manufacturer's CCS and should include a specific disinfection and monitoring programme approved by quality assurance. |
(続き) ・Pass-through hatches should be designed to protect the higher-grade environment, for example by effective flushing with an active filtered air supply ・The movement of material or equipment from lower grade or unclassified area to higher-grade clean areas should be subject to cleaning and disinfection commensurate with the risk and in line with the CCS. |
4.13 For pass-through hatches and airlocks (for material and personnel), the entry and exit doors should not be opened simultaneously. For airlocks leading to the grade A and grade B areas, an interlocking system should be used. For airlocks leading to grade C and D areas, a visual and/or audible warning system should be operated as a minimum. Where required to maintain area segregation, a time delay between the closing and opening of interlocked doors should be established. | 4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and/or an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of different grades should have an air pressure difference of a minimum of 10 Pascals (guidance value). Particular attention should be paid to the protection of the critical zone. The recommendations regarding air supplies and pressures may need to be modified where it is necessary to contain certain materials (e.g. pathogenic, highly toxic or radioactive products or live viral or bacterial materials). The modification may include positively or negatively pressurized airlocks that prevent the hazardous material from contaminating surrounding areas. | (続き) Decontamination of facilities (e.g. the cleanrooms and the heating, ventilation, and air conditioning (HVAC) systems) and the treatment of air leaving a clean area, may be necessary for some operations. Where containment requires air to flow into a critical zone, the source of the air should be from an area of the same or higher grade. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
PREMISES AND EQUIPMENT 3. Where products or clean components are exposed, the area should be fed with filtered air, should comply with the requirements of at least a Grade D environment and should be entered through airlocks. |
Glossary airlock airlock. An enclosed space with two or more doors, which is interposed between two or more rooms, e.g. of differing classes of cleanliness, for the purpose of controlling the airflow between those rooms when they need to be entered. An airlock is designed for use either by people or for goods and/or equipment. |
3. Glossary airlock An enclosed space with two or more doors, which is interposed between two or more rooms, e.g. of differing classes of cleanliness, for the purpose of controlling the airflow between those rooms when they need to be entered. An airlock is designed for and used by either people or goods (this can be a personnel airlock (PAL) or a material airlock (MAL)). |
8. Facility layout 8.2 The link between the interior and exterior of the premises should be through airlocks (PAL and/or MAL), changing rooms, pass boxes, passthrough hatches, decontamination devices, etc. These entry and exit doors for materials and personnel should have an interlock mechanism or other appropriate system to prevent the opening of more than one door at a time. |
9. Air-handling systems 9.3 Facilities and premises dealing with hazardous substances should have the following basic air-handling characteristics: ・ Airlocks, pass-through hatches, etc., should have supply and extract air to provide the necessary air pressure cascade and containment. The final, or containment perimeter, airlock or pass-through hatch bordering on an external or non-GMP area should be at a positive pressure relative to the environment, to prevent the ingress of contaminants to the facility. |
12. Personnel decontamination systems 12.2 An air shower comprises an airlock where high velocity air is supplied through air nozzles (e.g. from the sides of the airlock) in order to dislodge dust particles. Air extraction grilles (e.g. at low level) should draw the air away and return it to the filtration system. Some air showers may also incorporate a vertical unidirectional airflow section at the exit end, to flush contaminants away. |
4. Premises 4.9 Containment, cleanliness and protection may be facilitated through, for example: ・correct building layout;building finishes; ・the use of airlocks such as personnel airlocks (PAL) and/or material airlocks (MAL); ・pass-through hatches (PTH); ・change rooms and passages; ・sufficient pressure differentials. 4.13 Where appropriate, such as where the simultaneous opening of airlock doors might lead to a cross-contamination risk, airlock doors should not be opened at the same time. In such cases, controls such as interlocking systems, warning systems and procedures should be implemented. |
7. Air filtration, airflow direction and pressure differentials 7.11 Where airlocks are used, the pressure differentials selected should be appropriate. When selecting room pressure differentials, transient variations, such as machine extract systems and their impact, should be taken into consideration. |
3. Glossary airlock. An enclosed space with two or more doors, which is interposed between two or more rooms, for example, of differing classes of cleanliness, for the purpose of controlling the airflow between those rooms when they need to be entered. An airlock is designed for and used by either people or goods (personnel airlock (PAL); material airlock (MAL)). |
3. Glossary pass-through hatch or pass box. A cabinet with two or more doors for passing equipment, material or product while maintaining the pressure cascade and segregation between two controlled zones. A passive pass-through hatch (PTH) has no air supply or air extraction. A dynamic PTH has an air supply into the chamber. |
4. Premises 4.1 Premises design (略) The infiltration of contamination from outside air should be minimized by the use of appropriate filtration, room pressure differentials and airlocks. |
(続き)(略) Where necessary, air locks, change rooms and pass-through hatches may be considered and provided with effective ventilation and filtered air. Special attention should be given to door design, as gaps between doors and floors, doors opening into low-pressure areas, and sliding doors can result in changes in the pressure differential between areas. An interlocking system and a visual and/or audible warning system may be used, where required, to prevent opening of more than one door at a time where required. |
4.2 Weighing/dispensing and sampling areas A room for weighing (e.g. dispensing of materials) should be of appropriate design (for examples, see Figs A2.1 and A2.2). It is often advantageous to have several rooms associated with the weighing activity. These may include a preweighing staging area, personnel airlock, material airlock, weighing area with a containment booth, post-weighing staging area, washing area and provision for waste removal. |
(続き)(略) To further support containment, consideration may also be given to having material airlocks (MALs) and personnel airlocks (PALs), where needed, for entry and exit of processing areas (for an example, see Fig. A2.6). Appropriately designed airlocks can assist in ensuring containment. Additional controls, such as pressure differentials between areas, an appropriate number of air changes in an area, and sufficient filtration of air, should be in place. The use of airlocks assists in ensuring containment; however, other means may be considered to achieve this objective, such as closed systems and pressure gradients between adjacent areas. |
5.1 Containment Manufacturers should ensure that appropriate measures are taken to contain product dust in a manufacturing area, thus preventing or minimizing the risk of contamination of other areas and possible cross-contamination. In some cases, it may be advisable to have airlocks or pass-through hatches between rooms or areas. In addition, sufficient dilution, pressure differentials (recommended minimum values of 5 Pa) and airflow directions can further support containment in an area. |
4. Premises 4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to which should be through change rooms that act as airlocks. Cleanrooms and change rooms should be maintained at an appropriate cleanliness standard and supplied with air that has passed through filters of an appropriate efficiency. Controls and monitoring should be scientifically justified and should effectively evaluate the state of environmental conditions of cleanrooms, airlocks and pass- hrough hatches. |
4.12 Airlocks should be designed and used to provide physical separation and to minimize microbial and particle contamination of the different areas, and should be present for material and personnel moving between different grades. Wherever possible, airlocks used for personnel movement should be separated from those used for material movement. Where this is not practical, time-based separation of movement (personnel or material) by procedure should be considered. | (続き) Airlocks should be effectively flushed with filtered air to ensure that the grade of the cleanroom is maintained. The final airlock should, in the at rest state, be of the same cleanliness grade (viable and total particle) as the cleanroom into which it leads. The use of separate change rooms for entering and leaving the grade B area is desirable. Where this is not practical, time-based separation of activities (inward or outward) by procedure should be considered. Where the CCS indicates that the risk of contamination is high, separate change rooms for entering and leaving production areas should be used. |
(続き) Airlocks should be designed as follows: i.Personnel airlocks: areas of increasing cleanliness used for entry of personnel (for example, from the grade D area to the grade C area to the grade B area). In general, handwashing facilities should be provided only in the first change room and should not be present in change rooms directly accessing the grade B area. ii.Material airlocks: used for materials and equipment transfer. -Only materials and equipment that have been included on an approved list and assessed during validation of the transfer process should be transferred into the grade A or B areas via an airlock or pass-through hatch. |
(続き) Equipment and materials intended for use in the grade A area should be protected when transiting through the grade B area. Any unapproved items that require transfer should be preapproved as an exception. Appropriate risk assessment and mitigation measures should be applied and recorded as per the manufacturer’s CCS and should include a specific disinfection and monitoring programme approved by quality assurance. |
(続き) -Pass-through hatches should be designed to protect the highergrade environment, for example by effective flushing with active filtered air supply of appropriate grade in accordance with the CCS. -The movement of material or equipment from lower-grade or unclassified areas to higher-grade clean areas should be subject to cleaning and disinfection commensurate with the risk and in line with the CCS. |
4.13 For pass-through hatches and airlocks (for material and personnel), the entry and exit doors should not be opened simultaneously. For airlocks leading to the grade A and B areas, an interlocking system should be used. For airlocks leading to grade C and D areas, a visual or audible warning system should be operated as a minimum. Where required to maintain area segregation, a time delay between the closing and opening of interlocked doors should be established and validated. | 4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure and an airflow relative to the background environment of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of different grades should have an air pressure differential of a minimum of 10 pascals (guidance value). Particular attention should be paid to the protection of the critical zone. The recommendations regarding air supplies and air pressures may need to be modified where it is necessary to contain certain materials (such as pathogenic, highly toxic or radioactive products or live viral or bacterial materials). The modification may include positively or negatively pressurized airlocks that prevent the hazardous material from contaminating surrounding areas. | (続き) Decontamination (for example, of the cleanrooms and the heating, ventilation and air-conditioning (HVAC) systems) and the treatment of air leaving a clean area may be necessary for some operations. Where containment requires air to flow into a critical zone, the source of the air should be an area of the same or higher grade. |
Glossary Airlock - An enclosed space with interlocked doors, constructed to maintain air pressure control between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock is to preclude ingress of particle matter and microorganism contamination from a lesser controlled area. |
Glossary Pass-through hatch - Synonymous with airlock (see airlock definition) but typically smaller in size. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・直接支援区域と直接支援区域に隣接する区域とはエアロックにより分離すること。 ・エアロック扉には、同時開放を防止する機械式、電気式、目視、音の方法等を装置を備えること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・更衣室は、エアロックの機能を有すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・エアロックは、室間差圧及び気流の逆転が起きないよう、十分な差圧を設けること。 ・扉を閉じた状態で10〜15Pa又はそれ以上の差圧を維持すること。 ・清浄度レベルの異なる区域の間のエアロックは、適切な差圧を設けること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・直接支援区域と直接支援区域に隣接するその他の区域の清浄度が異なる場合は、エアロックにより分離すること。 ・同じ清浄度の場合は、それぞれの区域での作業内容による影響を勘案しエアロックを設けること。 ・エアロック扉には、同時開放を防止する機械式、電気式、目視、音の方法等を装置を備えること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・更衣室は、エアロックの機能を有すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・人は、エアロックとして機能する更衣室を経由してクリーンルームに入ること。 ・設備や原料は、エアロックを経由してクリーンルームに入れること。 ・エアロック、パススルーハッチの環境状態をが適切に評価すること。 *用語についての補足:「パススルーハッチ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの異なる等級間の微生物や粒子汚染を最小化するように、エアロックを設計し、使用すること。 ・クリーンルームの異なる等級間をエアロックで物理的な分離し、移動する人や原料用に、設置すること。 ・人用、物用のエアロックは、可能な限り、分離すること。分離が難しい場合は、時間の管理についての手順による検討を行うこと。 *用語についての補足:「エアロック」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・エアロックは、クリーンルーム等級を維持するために、ろ過空気で効果的にする置換すること。 ・エアロックは最終的に「at rest」状態で、それにつながるクリーンルームと同じ清浄度等級(生菌と粒子数)であること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・人用のエアロックは、グレードDからC、CからBのように清浄度は段階的に高くなるように設計すること。 ・原料や設備は、グレードA又はグレードBの区域内へエアロック又はパススルーハッチを通してを搬入すること。原料や設備は承認済で、搬送工程のバリデーションの際に評価済みであること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 *用語についての補足:「パススルーハッチ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・グレードA区域内用の物は、グレードB区域を通過する際に保護されていること。 ・承認済でない物の搬送は、例外として事前承認を受けること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・パススルーハッチは、より高いクリーンルーム等級を維持するために、ろ過空気で効果的にする置換すること。 *用語についての補足:「パススルーハッチ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・パススルーハッチやエアロック(物用と人用)は、出口と入口のドアを同時に開けないこととし、グレードAやグレードBの区域に通じるエアロックにはインターロックシステム、グレードCやDの区域に通じるエアロックには視覚的、聴覚的な警報システムを最低限、作動させること。 ・区域の隔離の維持が必要な場合、インターロックドアの閉鎖と開放に時間差を設定すること。 *グレードの表記は、GMP構造設備要求比較 空気調和設備 003:クリーンルーム清浄度の設定を参照ください。 *用語についての補足:「パススルーハッチ」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・封じ込めが必要な場合において、給気及び気圧に関する推奨事項の修正する場合がある。修正には、危険物質が周囲の区域を汚染するのを防止する陽圧又は陰圧のエアロックが含まれる場合がある。 *用語についての補足:「エアロック」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 封じ込めで重要区域に空気を流入場合には、当該空気の供給源は、同等以上のクリーンルーム等級の区域からとすること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・エアロックの定義。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・パススルーハッチの定義。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 以下の要件があります。 ・無菌工程における製造区域へ耐熱性のない物品及び原材料の搬入は、インターロックドア付きのエアロックを通じて搬入すること。 ・エアロックの扉は、所定の手順で表面を効果的に殺菌すること。 ・当該物品及び原材料について、清浄区域への搬入段階の数に応じて、幾重にも包装され、適切な表面消毒の予防策を講じた上で、エアロックを通して搬入すること。 |
対象医薬品:定量噴霧式吸入剤 以下の要件があります。 ・製品又は洗浄済みの構成部品が暴露される区域は、少なくともグレードのD環境の要求を満たし、エアロックを介して入室すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・エアロック及びインターロック扉(2重扉で片方が開いている間は、もう一方の扉はロックされるタイプ)は、空調バランス制御を容易にする。 ・エアロックは一般的に隣り合った異なる清浄度設定の室間に設けること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・エアロックの定義 ・隣接する室間(一般的に、クリーンルーム等級が異なる室間)で室の圧力を制御するために設けられた、インターロック扉付きの小部屋とのこと。 ・管理レベルの低い区域から粒子や微生物による汚染を防ぐとのこと。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.13と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品( 原薬及び製剤) の製造 パートA 一般的ガイダンス 作業原則 50.と同じ内容となります。 |
対象医薬品:定量噴霧式吸入剤 別紙(9) PIC/S GMP ガイドライン アネックス10 定量噴霧式吸入剤の製造 建物及び設備 3.と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.13と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
対象医薬品:定量噴霧式吸入剤 別紙(9) PIC/S GMP ガイドライン アネックス10 定量噴霧式吸入剤の製造 建物及び設備 3.と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・エアロックの定義 ・クリーンルーム等級が異なるなどの2室以上の部屋の間に設けられ、2枚以上の扉により閉じることができる空間のこと。 ・入室動作時にこれらの部屋間の気流を制御することを目的とする。 ・物/設備用と人用のエアロックが設計される。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・エアロックの定義 ・クリーンルーム等級が異なるなどの2室以上の部屋の間に設けられ、2枚以上の扉により閉じることができる空間のこと。 ・入室動作時にこれらの部屋間の気流を制御することを目的とする。 ・物用(MAL)と人用(PAL)のエアロックが設計される。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・施設の内部と外部の接続にはエアロック、更衣室、パスボックス、除染機器等を通すこと。 ・原料及び作業員が出入りする扉はインターロック機能もしくは他の一度に2ヶ所以上が開かないようにする適切なシステムを持つこと。 *用語ついての補足 パススルーハッチはパスボックスの意味である。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・ハザード物質を扱う場合、エアロック、パスボックス等は給気と排気を行い必要な室間差圧カスケードを作り封じ込めをおこなうこと。 ・最終の境界あるいは封じ込めの境界において、外部あるいは非GMPエリアに面するエアロック、パスボックスは周囲より陽圧にすることで施設への汚染物質の浸入を防ぐこと。 *用語ついての補足 パススルーハッチはパスボックスの意味である。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・作業員の徐塵について、エアシャワーは塵埃粒子を除去するためにノズルから高速の気流を(側面等から)吹き付ける機能を持つエアロックである。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・封じ込めや清浄化、防護のための設備例として、物用(MAL)と人用(PAL)のエアロック、パススルーハッチ等がある。 ・交差汚染のリスクがある場合にはエアロックの扉が同時が開かないようにすること。インターロック、警報、管理手順を持つこと。 *用語についての補足:「エアロック」「パススルーハッチ」は本表WHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・エアロックは、機械の排気システムなどによる一時的な変動やその影響を考慮し、圧力差が適切であること。 *用語についての補足:「エアロックは本表WHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・エアロックの定義。 ・エアロックは人用PALと物用MALを分けて設置する。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・パススルーハッチの定義。 ・パススルーハッチまたはパスボックスには換気を行わないタイプ(A passive PTH)と、給気を行うタイプ(A dynamic PTH)がある、 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・エアロックの設置により、外気からの汚染の侵入を抑えること。 *用語ついての補足:「エアロックは本表WHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・エアロックの設置により、効果的な換気とろ過された空気を供給する。 ・必要の応じて、複数のドアの同時開放を防止するために、インターロック、視覚的/聴覚的な警報を持つこと。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・秤量作業に関連する複数の部屋に、物用と人用のエアロックが含まれる。 *用語ついての補足:「エアロックは本表WHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・封じ込めを強化するために、物用(MAL)と人用(PAL)のエアロックを設けること。 ・エアロックの適切な設計は、封じ込めの確保に有用である。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・他のエリアへの汚染や交差汚染のリスクを防止するために、場合によって、室やエリア間にエアロックやパススルーハッチを設けることが望ましい。 *用語ついての補足:「パススルーハッチ」は本表WHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.1と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.12と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.13と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.14と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 エアロックと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 パススルーハッチと同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | 米国 | ー | ー | 欧州 | 欧州 | 欧州 | ー | ー | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CDER | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | |||||
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | Code of Federal Regulations | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||
| 法規類 タイトル |
薬局等構造設備規則 | 無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(5) PIC/S GMPガイドライン アネックス6 医療用ガスの製造 |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing ― Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 6 MANUFACTURE OF MEDICINAL GASES |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 6 Manufacture of Medicinal Gases |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, 986, 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||||
| 発行年 | 2021年 | 2011年 | 2012年 | 2020年 | 2025年 | 2013年 | 2023年 | 2004年 | 2023年 | 2023年 | 2015年 | 2022年 | 2010年 | 2014年 | 2022年 | |||||
| 記載内容載内容 | 第二章 医薬品等の製造業 第一節 医薬品の製造業 (無菌医薬品区分の医薬品製造業者等の製造所の構造設備) 第七条 施行規則第二十五条第一項第三号の区分及び施行規則第三十五条第一項第三号の区分の医薬品製造業者等の製造所の構造設備の基準は、前条に定めるもののほか、次のとおりとする。 二 無菌原薬に係る製品の作業所のうち、滅菌のために行う調製作業以後の作業の作業室(調製条件によつて菌の増殖を抑制できる場合を除く。)及び無菌医薬品(無菌原薬を除く。)に係る製品の作業所のうち、薬剤の調製作業、充填作業又は閉塞作業を行う作業室又は作業管理区域は、次に定めるところに適合するものであること。 イ 天井、壁及び床の表面は、消毒液等による噴霧洗浄に耐えるものであること。 |
(特定生物由来医薬品等の医薬品製造業者等の製造所の構造設備) 第八条 (中略)(以下「特定生物由来医薬品等」と総称する。)に係る製品の医薬品製造業者等の製造所の構造設備の基準は、第六条(無菌医薬品に係る製品の製造を行う場合においては、前二条)に定めるもののほか、次のとおりとする。 一 特定生物由来医薬品等に係る製品の製造所(包装、表示又は保管のみを行う製造所を除く。)は、次に定めるところに適合するものであること。 |
イ 清浄区域(作業所のうち、原料の秤量作業を行う場所、薬剤の調製作業を行う場所及び洗浄後の容器が作業所内の空気に触れる場所をいう。以下この号において同じ。)及び無菌区域(作業所のうち、無菌化された薬剤又は滅菌された容器が作業所内の空気に触れる場所、薬剤の充填作業を行う場所、容器の閉塞作業を行う場所及び無菌試験等の無菌操作を行う場所をいう。以下この号において同じ。)は、次に定めるところに適合するものであること。 (1) 天井、壁及び床の表面は、なめらかでひび割れがなく、かつ、じんあいを発生しないものであること。また、清掃が容易で、消毒を行うことができるものであること。 |
(放射性医薬品区分の医薬品製造業者等の製造所の構造設備) 第九条 施行規則第二十五条第一項第二号の区分及び施行規則第三十五条第一項第二号の区分の医薬品製造業者等の製造所(包装、表示又は保管のみを行う製造所を除く。以下この項及び次項において同じ。)の構造設備の基準は、第六条及び第七条に定めるもののほか、次のとおりとする。 二 放射性医薬品に係る製品の作業所は、次に定めるところに適合するものであること。 ホ 次に定めるところに適合する作業室及び試験検査室(動物試験を行う場合には動物試験室を含む。以下同じ。)を有すること。 |
(1) 内部の壁、床その他放射性物質(放射性医薬品の製造及び取扱規則第一条第二号に規定する放射性物質をいう。以下同じ。)によつて汚染されるおそれのある部分は、突起物、くぼみ及び仕上げ材の目地等のすきまの少ない構造であること。 (2) 内部の壁、床その他放射性物質によつて汚染されるおそれのある部分の表面は、平滑であり、気体又は液体が浸透しにくく、かつ、腐食しにくい材料で仕上げられていること。 |
6. 構造設備 6.1 構造設備の設計上の要点 6) 天井は効果的にシールされていること。 16) 壁、床及び天井の表面は、清掃可能で洗浄剤や消毒剤に耐える材質であること。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 6) 天井は、効果的にシールされていること。 14) 壁、床及び天井の表面は、清掃可能で洗浄剤や消毒剤に耐える材質であること。 |
第3章 建物及び設備 建物 製造区域 3.9. 出発原料及び一次包装材料、中間製品又はバルク製品が露出する環境においては、内装表面(壁、床及び天井)が、平滑でひび割れ及び開放接合部がなく、微粒子物質を脱落させないものであるとともに、容易かつ効果的な清浄化及び(必要であれば)消毒を行えるものであること。 |
4 建物 4.7 クリーンルーム内で用いられる材質(当該室の構造建材及び当該室内部で使用される物品の材質の両方)は、微粒子の発生が最小化されるように、また、清浄化剤、消毒剤及び殺芽胞剤を使用する場合には、それらを繰り返し適用できるように、選定されていること。 4.8 天井は、上部空間からの汚染を防止するように設計され且つ閉塞されていること。 |
建物と設備 建物 8. 建物は、混同のリスクを避けるため、製造、試験、貯蔵工程を行なうのに充分なスペースを備えていること。建物は下記のように定めること。 (略) b) それぞれの製造工程の段階のシリンダー/移動型極低温容器は識別、隔離を明確にする。(例えば「検査待ち」、「充てん待ち」、「判定待ち」、「検査済み」、「不合格」、「出荷待ち」) これらの様々なレベルの隔離を達成するために用いられる方法は、全体の作業の性質、範囲、複雑さによって決まる。床面への表示、間仕切り、仕切り、標識、ラベル、他の適切な方法が用いられる。 |
Subpart C - Buildings and Facilities Sec. 211.42 Design and construction features. (c) Operations shall be performed within specifically defined areas of adequate size. There shall be separate or defined areas or such other control systems for the firm's operations as are necessary to prevent contamination or mixups during the course of the following procedures: (10) Aseptic processing, which includes as appropriate: (i) Floors, walls, and ceilings of smooth, hard surfaces that are easily cleanable; |
IV. BUILDINGS AND FACILITIES 21 CFR 211.42(c) states, in part, that “Operations shall be performed within specifically defined areas of adequate size. There shall be separate or defined areas or such other control systems for the firm’s operations as are necessary to prevent contamination or mixups during the course of the following procedures: (10) Aseptic processing, which includes as appropriate: (i) Floors, walls, and ceilings of smooth, hard surfaces that are easily cleanable; (略) |
E. Design Cleanrooms are normally designed as functional units with specific purposes. The materials of construction of cleanrooms ensure ease of cleaning and sanitizing. Examples of adequate design features include seamless and rounded floor to wall junctions as well as readily accessible corners. Floors, walls, and ceilings should be constructed of smooth, hard surfaces that can be easily cleaned. Ceilings and associated HEPA filter banks should be designed to protect sterile materials from contamination. Cleanrooms also should not contain unnecessary equipment, fixtures, or materials. |
4 Premises 4.7 Materials used in cleanrooms, both in the construction of the room and for items used within the room, should be selected to minimize generation of particles and to permit the repeated application of cleaning, disinfectant and sporicidal agents where used. 4.8 Ceilings should be designed and sealed to prevent contamination from the space above them. |
PREMISES AND EQUIPMENT Premises 8. Premises should provide sufficient space for manufacturing, testing and storage operations to avoid the risk of mix-up. Premises should be designated to provide: (略) b) clear identification and segregation of cylinders/mobile cryogenic vessels at various stages of processing (e.g. “waiting checking”, ""awaiting filling"", ""quarantine"", ""certified"", “rejected “,“prepared deliveries”). The method used to achieve these various levels of segregation will depend on the nature, extent and complexity of the overall operation. Marked-out floor areas, partitions, barriers, signs, labels or other appropriate means could be used. |
CHAPTER 3 PREMISES AND EQUIPMENT Premises Production Area 3.9 Where starting and primary packaging materials, intermediate or bulk products are exposed to the environment, interior surfaces (walls, floors and ceilings) should be smooth, free from cracks and open joints, and should not shed particulate matter and should permit easy and effective cleaning and, if necessary, disinfection. |
4 Premises 4.7 Materials used in cleanrooms, both in the construction of the room and for items used within the room, should be selected to minimize generation of particles and to permit the repeated application of cleaning, disinfectant and sporicidal agents where used. 4.8 Ceilings should be designed and sealed to prevent contamination from the space above them. |
Premises and equipment Premises 8. Premises should provide sufficient space for manufacturing, testing and storage operations in order to prevent any risk of mix-up. Premises should be designated to provide: (略) b) clear identification and segregation of cylinders/mobile cryogenic vessels at various stages of processing (e.g. “waiting checking”, ""awaiting filling"", ""quarantine"", ""certified"", “rejected “,“prepared deliveries”). The method used to achieve these various levels of segregation will depend on the nature, extent and complexity of the overall operation. Marked-out floor areas, partitions, barriers, signs, labels or other appropriate means could be used. |
12. Premises Production areas 12.27 Where starting and primary packaging materials and intermediate or bulk products are exposed to the environment, interior surfaces (walls, floors and ceilings) should be smooth and free from cracks and open joints, should not shed particulate matter, and should permit easy and effective cleaning and, if necessary, disinfection. |
4. Premises 4.7 Materials used in cleanrooms, both in the construction of the room and for items used within the room, should be selected to minimize generation of particles. These should permit the repeated application of cleaning, disinfecting and sporicidal agents where used. 4.8 Ceilings should be designed and sealed to prevent contamination from the space above them. |
| 概 要 | 対象医薬品:無菌医薬品 以下の要件があります。 ・天井、壁及び床の表面は、消毒液等による噴霧洗浄に耐えるものであること。 |
ー | 対象医薬品:特定生物由来医薬品 以下の要件があります。 ・天井、壁及び床の表面は、なめらかでひび割れがなく、塵埃を発生しないものであること。 ・清掃が容易で、消毒を行えることができるものであること。 |
ー | 対象医薬品:放射性医薬品 以下の要件があります。 ・内部の壁、床、その他放射性物質によって汚染される恐れのある部分は、突起物、くぼみ及び仕上材の目地等の隙間の少ない構造であること。 ・内部の壁、床、その他放射性物質によって汚染される恐れのある部分の表面は、平滑であり、機体または液体が浸透しにくくかつ、腐食しにくい材料で仕上げられていること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・天井は効果的にシールされていること。 ・壁、床及び天井の表面は、清掃可能で洗浄剤や消毒剤に耐える材質であること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・天井は効果的にシールされていること。 ・壁、床及び天井の表面は、清掃可能で洗浄剤や消毒剤に耐える材質であること。 |
対象医薬品: 全般 以下の要件があります。 ・出発原料及び一次包装材料、中間製品又はバルク製品が露出される環境では、建物内部の表面(壁・床及び天井)は平滑で、隙間がなく、また微粒子物質を脱落させず、また容易かつ効果的な清掃、及び必要な場合は消毒が行えるものであること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルームの建材は、微粒子の発生が最小化されるものであること。 ・クリーンルームの建材は、清清浄化剤、消毒剤及び殺芽胞剤を使用する場合、繰り返し使用に適用できるものであること。 ・天井は、その上の空間からの汚染を防ぐように設計し、シールすること。 *用語ついての補足:閉塞とは、”仕舞のシール”の意味である。 |
対象医薬品:医療用ガス 以下の要件があります。 ・それぞれの製造工程の段階のシリンダー/移動型極低温容器は識別、隔離を明確にする必要があるが、床面への表示、他の適切な方法が用いられる。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・床、壁及び天井は容易に清掃ができるように平滑であること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・「21 CFR211.42 (c)(10) (i)」 では、床、壁及び天井は容易に清掃ができるように平滑であること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・クリーンルームの建材は、清掃や消毒が容易であること。 ・床と壁の接合部が継ぎ目なく丸みを帯び、隅が簡単にアクセスできること。 ・床、壁、天井は、滑らかで硬い表面で構成されており、容易に清掃できること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.7、4.8と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 建物と設備 建物 8. b)と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 製造区域 3.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.7、4.8と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 建物と設備 建物 8. b)と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 製造区域 3.9と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.7、4.8と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | ー | ー | ー | ー | ー | ー | 欧州 | 欧州 | 欧州 | 欧州 | 欧州 | 欧州 | ー | ー | |||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | FDA | PIC/S | PIC/S | PIC/S | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | |||||||||||||||||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長 | Code of Federal Regulations | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||||||||||||||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 最終滅菌法による無菌医薬品の製造に関する指針 | 医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令の一部改正について | PIC/SのGMPガイドラインを活用する際の考え方について 別紙(1) PIC/S GMP ガイドライン パート 1 |
PIC/SのGMPガイドラインを活用する際の考え方について 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
「PIC/S のGMP ガイドラインを活用する際の考え方について」の一部改正について 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 |
「PIC/S のGMP ガイドラインを活用する際の考え方について」の一部改正について 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性医薬品の製造 |
「PIC/S のGMP ガイドラインを活用する際の考え方について」の一部改正について 別紙(12) PIC/S GMP ガイドライン アネックス13 治験薬の製造 |
「PIC/S のGMP ガイドラインを活用する際の考え方について」の一部改正について 別紙(13) アネックス14 ヒト血液及びヒト血漿由来製品の製造 |
原薬GMPのガイドラインについて | TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER F--BIOLOGICS PART 600 BIOLOGICAL PRODUCTS: GENERAL9 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 3 MANUFACTURE OF RADIOPHARMACEUTICALS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 6 MANUFACTURE OF MEDICINAL GASES |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 7 MANUFACTURE OF HERBAL MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 13 MANUFACTURE OF INVESTIGATIONAL MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 14 MANUFACTURE OF MEDICINAL PRODUCTS DERIVED FROM HUMAN BLOOD OR PLASMA |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 3 Manufacture of Radiopharmaceuticals |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 6 Manufacture of Medicinal Gases |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 7 Manufacture of Herbal Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 13 Investigational Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 14 Manufacture of Medicinal Products Derived from |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 45th report Annex 2 WHO good practices for pharmaceutical microbiology laboratories WHO Technical Report Series, No. 961, 2011 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
|||||||||||||||||||||||||||||
| 発行年 | 2011年 | 2012年 | 2021年 | 2022年 | 2012年 | 2013年 | 2013年 | 2013年 | 2015年 | 2001年 | 2023年 | 2021年 | 2023年 | 2023年 | 2023年 | 2023年 | 2023年 | 2015年 | 2008年 | 2010年 | 2009年 | 2010年 | 2011年 | 2014年 | 2019年 | |||||||||||||||||||||||||||||
| 記載内容 |
6. 構造設備 6.1 構造設備の設計上の要点 28)更衣室は、作業する部屋の清浄度に合わせ適切に設けること。同じ清浄度でも原材料や製品などへの交叉汚染のリスクがある場合にはリスクに応じて追加の更衣室を設けることが望ましい。 |
5. 建物及び施設 5.1 構造設備の設計上の要点 13) 生理活性の高い物質や病原性物質、高毒性物質、放射性物質等を取り扱う場合には、交叉汚染等のリスクに応じた適切な構造設備を考慮すること。 27) 更衣室は、作業する部屋の清浄度に合わせ適切に設けること。同じ清浄度内でも原材料や製品などへの交叉汚染のリスクがある場合には、リスクに応じて個別の更衣室を設けること。 |
第2章医薬品製造業者等の製造所における製造管理及び品質管理 医薬品の製造業者等の製造所における製造管理及び品質管理について規定するものであること。 第1節通則 13.第9条(構造設備)関係 (1)医薬品に係る製品の製造所の構造設備に関して、その製造所における製造工程等に応じて要否を判断する事項を規定するものであり、それらに鑑みて製造所の構造設備が適合しているかどうかを判断するものであること。 ⑤第9条第1項第5号関係 医薬品に係る製品等を取り扱う作業室(密閉容器に納められた製品等のみを取り扱う作業室及び製品等から採取された検体のみを取り扱う作業室を除く。)に関して、当該製品等に専用とすること及び当該製品の漏出を防止する措置(いわゆる封じ込め措置)を要する場合について規定するものであること。 |
(続き)(略) ウ.交叉汚染することにより他の製品等に重大な影響が及ぶ製品等として、例えば、細胞毒性を有する抗がん剤等の劇薬又は毒薬のように、強い薬理作用又は毒性を有する製品等を含むものであること。当該製品等を取り扱う作業室において、交叉汚染を防止する適切な措置をとることができない場合には、当該作業室を当該製品等に専用とするとともに、当該製品等の漏出を防止する措置(いわゆる封じ込め措置)を要するものであること。 |
第3章 建物及び設備 原則 実施される作業にふさわしいように、建物及び装置を配置し、設計し、 建造し、供用し、保守管理しなければならない。その配置及び設計は、過誤のリスクを最小にすることを目途とするとともに、交叉汚染、じん埃又は汚れの蓄積及び(一般的に)製品品質への悪影響を回避するために、有効な洗浄及び 保守管理を可能とするものでなければならない。 |
製造区域 3.6. 交叉汚染による重篤な医学的危害のリスクを最小限にするため、高感作性の原材料(例えばペニシリン類)又は生物学的製剤(例えば生きている微生物に由来するもの)等の特殊な医薬品の製造には、専用化された自己封じ込め式の設備が利用可能でなければならない。ある種の抗生剤、ある種のホルモン、ある種の細胞毒性物質、ある種の高活性薬物及び非医薬品等の製品の製造は、同一の施設で実施してはならない。例外として、特別な予防策が講じられ、必要なバリデーションが行われている場合には、これら製品について同一施設におけるキャンペーン生産*訳注は許容され得る。工業毒物(殺虫剤及び除草剤等)の製造は、医薬品の製造に使用する建物では許されない 。 (*訳注:品目毎に時期を分けた集中生産を指す。 ) |
3.8. 異なる医薬品又はその構成物の混同を最小化し、交叉汚染を回避し、製造若しくは管理ステップの実施漏れ又は誤った適用のリスクを最小限にするよう、適切な作業スペース及び工程内保管スペースに、装置及び物品を整然と論理的に配置すること。 3.14. じん埃が発生する場合(例えば、サンプリング、秤量、混合及び加工の作業中、乾いた状態の製品の包装時)は、交叉汚染を回避して清掃を行いやすくする特別な予防措置を講じること。 3.15. 医薬品の包装のための建物は、混同又は 交叉汚染を回避できるよう、特別に設計し、配置すること。 保管区域 3.22. 通常、出発原料用に分離した検体採取区域があること。検体採取が保管区域で実施される場合は、汚染又は交叉汚染を防止するような方法で行うこと。 |
品質管理区域 3.27. 管理試験室は、そこで行われる作業に適するよう設計すること。混同及び交叉汚染を避けるため十分なスペースを与えること。検体及び記録書のための適切で相応の保管スペースがあること。 |
第5章 製造 包装作業 5.44. 包装作業のプログラムを設定する場合は、交叉汚染、混同又は取違いのリスクを最小化するため特別の注意を払うこと。物理的に隔離されていない限り、異なる製品を近接して包装してはならない。 |
建物及び設備 全般事項 17. 従業員、原材料、放射性核種などからの交叉汚染を予防する対策を立て、実施しなければならない。必要な場合には常に、閉鎖系装置又は封じ込め装置を用いなければならない。開放系装置を使用する場合、又は装置が開放されている場合は、汚染のおそれを最小限にするための予防措置を講じなければならない。リスク評価を行い、提案された環境清浄度レベルが、生産されている製品形態に適していることを実証しなければならない。 製造 34. 同じ作業区域(ホットセル、LAFユニットなど)での異なる放射性製品を同時に製造することは、交叉汚染や混同のリスクを最小限にするため避けなければならない。 |
原則 医療用ガスの製造 一般に医療用ガスの製造は閉鎖系の設備で行なわれる。従って、製品の環境からの汚染は極めて少ない。しかし、特に、容器の再利用によって、汚染(或いは他のガスとの交叉汚染)のリスクが生じる可能性がある。 |
設備 11. 設備は、確実に適正なガスが適正な容器に充てんされるように設計すること。通常、異なる種類のガスを移送するパイプライン間で交叉接続がないこと。交叉接続が必要な場合(例えば、混合物の充てん設備)、適格性評価により異なる種類のガスの交叉汚染のリスクがないことを保証すること。さらに、マニホールドは特定の接続を装備すること。これらの接続は、国際規格或いは国内規格に従うべきであろう。同一の充てん所での、別規格の接続の使用は、何らかの状況で特定の充てん接続システムに側管を通すために必要とするアダプターの使用と同様に、注意深く管理すること。 |
建物 保管区域 1. 植物薬は区分けされた場所に保管すること。保管区域は昆虫、或いはその他の動物、特に齧歯動物の侵入を防ぐことができるように設備を備えること。粗原料とともに運ばれるいかなる動物及び微生物の蔓延防止、発酵やカビの増殖及び交叉汚染を防止する効果的な措置を講じること。受入れた植物薬の隔離保管のため、及び合格した植物薬のために区分された場所を使用すること。 |
製造区域 5. 植物薬及び植物薬調製品の検体採取、秤量、混合及び加工作業を行う際に、粉塵が生じる場合は、例えば集塵装置や専用施設を使用するなど、容易に清掃ができ、交叉汚染が防止できるような具体的な対策を講じること。 |
建物及び設備 5. 毒性、効力(効能)、感作性は治験薬では完全に解明されておらず、そのため交叉汚染の全リスクを最小化することの必要性が強く求められる。設備や構造の設計、検査や試験方法及び洗浄後における許容限界についてはこれらリスクの特性を反映すること。キャンペーン製造については適切な考慮をすること。洗浄溶剤の選択に際しては治験薬の溶解性を考慮すること。 |
5. 構造設備 5.3 血漿由来医薬品の製造中に、適切なウイルス不活性化及び除去を用い、処理製品と未処理製品による交叉汚染を防止する手段を講じること。専用のかつ別個の施設及び装置をウイルス不活性化処理の前後の製造工程で使用すること。 |
4 構造及び設備 4.4 封じ込め 4.42 ある専用区域から別の専用区域へ移動する従業員、原材料等による交叉汚染を防止するため、適切な対策を確立し、実施すること。 5. 工程装置 5.2 装置の保守及び清掃 5.24 専用ではない装置については、交叉汚染を防止するため、異なる原薬等の製造の間に清掃すること。 |
7 原材料等の管理 7.2 受入及び区分保管 7.22 バルクが専用ではないタンクにより輸送される場合、タンクからの交叉汚染が発生しないことを保証すること。 その保証の手段としては、次の方法があり得る。 -洗浄済証明書 -微量不純物の試験 -供給業者の査察 7.4 保管 7.40 原材料等は、分解、汚染及び交叉汚染を防止するよう、取り扱い、保管すること。 |
16 受託製造業者(試験機関を含む) 16.10 全ての受託製造業者(試験機関を含む)は本ガイドラインで規定したGMPに従うこと。交叉汚染の防止及びトレーサビリティの維持に特別の考慮を払うこと。 17 代理店、仲介業者、貿易業者、流通業者、再包装業者及び再表示業者 17.4 原薬・中間体の再包装、再表示及び保管 17.41 再包装は、汚染及び交叉汚染を避けるために、適切な環境条件下で実施すること。 |
18 細胞培養・発酵により生産する原薬のガイドライン 18.3 細胞培養・発酵 18.38 共用装置(多品種製造)では、交叉汚染のリスクを最小限にするために、製品の一連の期間製造(キャンペーン製造)の間に、適切に、清掃後の追加試験が要求されることがある。 |
Subpart B--Establishment Standards Sec. 600.11 Physical establishment, equipment, animals, and care. (4) Live vaccine processing. Live vaccine processing must be performed under appropriate controls to prevent cross contamination of other products and other manufacturing areas within the building. Appropriate controls must include, at a minimum: (i)(A) Using a dedicated manufacturing area that is either in a separate building, in a separate wing of a building, or in quarters at the blind end of a corridor and includes adequate space and equipment for all processing steps up to, but not including, filling into final containers; and |
(続き) (B) Not conducting test procedures that potentially involve the presence of microorganisms other than the vaccine strains or the use of tissue culture cell lines other than primary cultures in space used for processing live vaccine; |
(続き) or (ii) If manufacturing is conducted in a multiproduct manufacturing building or area, using procedural controls, and where necessary, process containment. Process containment is deemed to be necessary unless procedural controls are sufficient to prevent cross contamination of other products and other manufacturing areas within the building. Process containment is a system designed to mechanically isolate equipment or an area that involves manufacturing using live vaccine organisms. A |
(続き) All product, equipment, and personnel movement between distinct live vaccine processing areas and between live vaccine processing areas and other manufacturing areas, up to, but not including, filling in final containers, must be conducted under conditions that will prevent cross contamination of other products and manufacturing areas within the building, including the introduction of live vaccine organisms into other areas. |
(続き) In addition, written procedures and effective processes must be in place to adequately remove or decontaminate live vaccine organisms from the manufacturing area and equipment for subsequent manufacture of other products. Written procedures must be in place for verification that processes to remove or decontaminate live vaccine organisms have been followed. |
(5) Equipment and supplies--contamination. Equipment and supplies used in work on or otherwise exposed to any pathogenic or potentially pathogenic agent shall be kept separated from equipment and supplies used in the manufacture of products to the extent necessary to prevent cross-contamination. | CHAPTER 3 PREMISES AND EQUIPMENT PRINCIPLE Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt and, in general, any adverse effect on the quality of products. |
Production Areas 3.6 Cross-contamination should be prevented for all products by appropriate design and operation of manufacturing facilities. The measures to prevent cross-contamination should be commensurate with the risks. Quality Risk Management principles should be used to assess and control the risks. Depending of the level of risk, it may be necessary to dedicate premises and equipment for manufacturing and/or packaging operations to control the risk presented by some medicinal products. |
3.8 The adequacy of the working and in-process storage space should permit the orderly and logical positioning of equipment and materials so as to minimise the risk of confusion between different medicinal products or their components, to avoid cross-contamination and to minimise the risk of omission or wrong application of any of the manufacturing or control steps. 3.14 In cases where dust is generated (e.g. during sampling, weighing, mixing and processing operations, packaging of dry products), specific provisions should be taken to avoid cross-contamination and facilitate cleaning. 3.15 Premises for the packaging of medicinal products should be specifically designed and laid out so as to avoid mix-ups or cross-contamination. |
Storage Areas 3.22 There should normally be a separate sampling area for starting materials. If sampling is performed in the storage area, it should be conducted in such a way as to prevent contamination or cross-contamination. Quality Control Areas 3.27 Control laboratories should be designed to suit the operations to be carried out in them. Sufficient space should be given to avoid mix-ups and cross-contamination. There should be adequate suitable storage space for samples and records. |
CHAPTER 5 PRODUCTION PACKAGING OPERATIONS 5.49 When setting up a programme for the packaging operations, particular attention should be given to minimising the risk of cross-contamination, mix-ups or substitutions. Different products should not be packaged in close proximity unless there is physical segregation. |
PREMISES AND EQUIPMENT General 17. Measures should be established and implemented to prevent cross-contamination from personnel, materials, radionuclides etc. Closed or contained equipment should be used whenever appropriate. Where open equipment is used, or equipment is opened, precautions should be taken to minimize the risk of contamination. The risk assessment should demonstrate that the environmental cleanliness level proposed is suitable for the type of product being manufactured. PRODUCTION 34. Production of different radioactive products in the same working area (i.e. hot-cell, LAF unit), at the same time should be avoided in order to minimise the risk of cross-contamination or mix-up. |
PRINCIPLE Manufacture of medicinal gases Manufacture of medicinal gases is generally carried out in closed equipment. Consequently, environmental contamination of the product is minimal. However, risks of contamination (or cross contamination with other gases) may arise, in particular because of the reuse of containers. |
Equipment 11. Equipment should be designed to ensure the correct gas is filled into the correct container. There should normally be no cross connections between pipelines carrying different gases. If cross connections are needed (e.g. filling equipment of mixtures), qualification should ensure that there is no risk of cross contamination between the different gases. In addition, the manifolds should be equipped with specific connections. These connections may be subject to international or national standards. The use of connections meeting different standards at the same filling site should be carefully controlled, as well as the use of adaptors needed in some situations to bypass the specific fill connection systems. |
PREMISES Storage areas 1. Herbal substances should be stored in separate areas. The storage area should be equipped in such a way as to give protection against the entry of insects or other animals, especially rodents. Effective measures should be taken to prevent the spread of any such animals and micro-organisms brought in with the crude substance, to prevent fermentation or mould growth and to prevent cross-contamination. Different enclosed areas should be used to quarantine incoming herbal substances and for the approved herbal substances. |
Production area 5. Specific provisions should be made during sampling, weighing, mixing and processing operations of herbal substances and herbal preparations whenever dust is generated, to facilitate cleaning and to avoid cross-contamination, as for example, dust extraction, dedicated premises, etc. |
4. PREMISES AND EQUIPMENT 1. The toxicity, potency or sensitising potential may not be fully understood for investigational medicinal products and this reinforces the need to minimise all risks of cross-contamination. The design of equipment and premises, inspection/test methods and acceptance limits to be used after cleaning should reflect the nature of these risks and take account of the quality risk management principles detailed in Chapters 3 and 5 of Part 1 of the PIC/S GMP Guide. |
5. PREMISES AND EQUIPMENT 5.3 In the production of plasma-derived medicinal products, appropriate viral inactivation or removal procedures are used and steps should be taken to prevent cross contamination of treated with untreated products. Dedicated and distinct premises and equipment should be used for manufacturing steps before and after viral inactivation treatment. |
PART I Chapter 3 Premises and Equipment PRINCIPLE Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt and, in general, any adverse effect on the quality of products. |
Production Areas 3.6 Cross-contamination should be prevented for all products by appropriate design and operation of manufacturing facilities. The measures to prevent cross-contamination should be commensurate with the risks. Quality Risk Management principles should be used to assess and control the risks. Depending of the level of risk, it may be necessary to dedicate premises and equipment for manufacturing and/or packaging operations to control the risk presented by some medicinal products. |
3.8 The adequacy of the working and in-process storage space should permit the orderly and logical positioning of equipment and materials so as to minimise the risk of confusion between different medicinal products or their components, to avoid cross-contamination and to minimise the risk of omission or wrong application of any of the manufacturing or control steps. 3.14 In cases where dust is generated (e.g. during sampling, weighing, mixing and processing operations, packaging of dry products), specific provisions should be taken to avoid cross-contamination and facilitate cleaning. 3.15 Premises for the packaging of medicinal products should be specifically designed and laid out so as to avoid mix-ups or cross-contamination. |
Storage Areas 3.22 There should normally be a separate sampling area for starting materials. If sampling is performed in the storage area, it should be conducted in such a way as to prevent contamination or cross-contamination. Quality Control Areas 3.27 Control laboratories should be designed to suit the operations to be carried out in them. Sufficient space should be given to avoid mix-ups and cross-contamination. There should be adequate suitable storage space for samples and records. |
CHAPTER 5 PRODUCTION PACKAGING OPERATIONS 5.49 When setting up a programme for the packaging operations, particular attention should be given to minimising the risk of cross-contamination, mix-ups or substitutions.Different products should not be packaged in close proximity unless there is physical segregation. |
PREMISES AND EQUIPMENT General 17. Measures should be established and implemented to prevent crosscontamination from personnel, materials, radionuclides etc. Closed or contained equipment should be used whenever appropriate. Where open equipment is used, or equipment is opened, precautions should be taken to minimize the risk of contamination. The risk assessment should demonstrate that the environmental cleanliness level proposed is suitable for the type of product being manufactured. PRODUCTION 34. Production of different radioactive products in the same working area (i.e. hotcell, LAF unit), at the same time should be avoided in order to minimise the risk of cross-contamination or mix-up. |
PRINCIPLE Manufacture of Medicinal Gases Manufacture of medicinal gases is generally carried out in closed equipment. Consequently, environmental contamination of the product is minimal. However, risks of contamination (or cross contamination with other gases) may arise, in particular because of the reuse of containers. |
Equipment 11. Equipment should be designed to ensure the correct gas is filled into the correct container. There should normally be no cross connections between pipelines carrying different gases. If cross connections are needed (e.g. filling equipment of mixtures), qualification should ensure that there is no risk of cross contamination between the different gases. In addition, the manifolds should be equipped with specific connections. These connections may be subject to national or international standards. The use of connections meeting different standards at the same filling site should be carefully controlled, as well as the use of adaptors needed in some situations to bypass the specific fill connection systems. |
Premises & Equipment Storage areas 1. Herbal substances should be stored in separate areas. The storage area should be equipped in such a way as to give protection against the entry of insects or other animals, especially rodents. Effective measures should be taken to prevent the spread of any such animals and micro-organisms brought in with the herbal substance, to prevent fermentation or mould growth and to prevent cross-contamination. Different enclosed areas shouldbe used to quarantine incoming herbal substances and for the approved herbal substances. |
Production area 5. Specific provisions should be made during sampling, weighing, mixing and processing operations of herbal substances and herbal preparations whenever dust is generated, to facilitate cleaning and to avoid cross-contamination, as for example, dust extraction, dedicated premises, etc. |
PREMISES AND EQUIPMENT 5. The toxicity, potency and sensitising potential may not be fully understood for investigational medicinal products and this reinforces the need to minimise all risks of cross-contamination. The design of equipment and premises, inspection / test methods and acceptance limits to be used after cleaning should reflect the nature of these risks. Consideration should be given to campaign working where appropriate. Account should be taken of the solubility of the product in decisions about the choice of cleaning solvent. |
Human Blood or Plasma 5. Premises and Equipment 5.3 In the production of plasma-derived medicinal products, appropriate viral inactivation or removal procedures are used and steps should be taken to prevent cross contamination of treated with untreated products. Dedicated and distinct premises and equipment should be used for manufacturing steps after viral inactivation treatment. |
12. Premises Storage areas 12.3 Where dust is generated (e.g. during sampling, weighing, mixing and processing operations, or packaging of powder), measures should be taken to avoid cross-contamination and facilitate cleaning. 12.22 There should normally be a separate sampling area for starting materials. (If sampling is performed in the storage area, it should be conducted in such a way as to prevent contamination or cross-contamination.) 12.31 Premises for the packaging of pharmaceutical products should be specifically designed and laid out so as to avoid mix ups, contamination or cross-contamination. |
13. Equipment 13.1 Equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. The layout and design of equipment must aim to minimize the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt, and, in general, any adverse effect on the quality of products. 13.12 Non-dedicated equipment should be cleaned according to validated cleaning procedures between being used for production of different pharmaceutical products to prevent cross-contamination. |
16. Good practices in production Prevention of cross-contamination and bacterial contamination during production 16.12 Cross-contamination should be avoided by taking appropriate technical or organizational measures, for example: (c) providing appropriately designed airlocks, pressure differentials, and air supply and extraction systems; (g) using a “closed system” in production; |
4. Premises 4.1 Premises design Both the architectural design of the building and that of the HVAC system should be carefully considered when attempting to achieve the general objectives of preventing contamination and cross-contamination and ensuring an appropriate environment for the production and control of pharmaceutical products. It is important to ensure that the required environmental conditions, cleanliness and containment are achieved and maintained. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・更衣室について交叉汚染のリスクに応じて追加の更衣室を設けること。 |
対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・交叉汚染等のリスクに応じた適切な構造設備を考慮すること。 ・更衣室について交叉汚染のリスクに応じて追加の更衣室を設けること。 |
ー | 対象医薬品:一般医薬品 以下の要件があります。 ・交叉汚染することにより他の製品等に重大な影響が及ぶ製品等の場合の専用作業室について。 |
対象医薬品: 全般 以下の要件があります。 ・建物及び装置は、交叉汚染等製品品質へのいかなる悪影響も回避するための有効な洗浄と保守管理を可能とすること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・同一の施設で、高感作性の原材料(例えばペニシリン類)又は生物学的製剤(例えば生きている微生物に由来するもの)等の特殊な医薬品が製造できない場合について。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・作業場所及び工程内保管場所は、装置及び原材料を整然と論理的に配置すること。 ・塵埃が発生する場合には、交叉汚染を回避し清掃を行いやすいように特別な予防措置が取られること。 ・包装のための建物は交叉汚染を回避できるように設計及び配置すること。 ・サンプリングが保管区域で行われる場合は、交叉汚染を防止するような方法で実施すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・管理試験室は、交叉汚染を避けるための十分なスペースが与えられていること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・包装作業ににおいては交叉汚染のリスクを最小とするため、物理的な隔離がない限り、異なる製品を近接して包装しないこと。 |
対象医薬品:放射性医薬品の製造 以下の要件があります。 ・従業員、原材料、放射性核種などからの交叉汚染を予防するリスク評価を含む対策を立て、実施すること。 ・同じ作業区域(ホットセル、LAFユニットなど)での異なる放射性製品を同時に製造することは避けること。 |
対象医薬品:医療用ガス 以下の要件があります。 ・医療用ガスの製造においては一般に閉鎖系の設備で行なわれるが、容器の再利用によって、汚染(或いは他のガスとの交叉汚染)のリスクを検討すること。 |
対象医薬品:医療用ガス 以下の要件があります。 ・容器に充てんされる際、異なる種類のガスを移送するパイプライン間で交叉接続がないこと。 ・同交叉接続が必要な場合、適格性評価により異なる種類のガスの交叉汚染のリスクがないことを保証すること。 |
対象医薬品:植物性医薬品 以下の要件があります。 ・保管区域は、粗原料とともに運ばれるいかなる動物及び微生物の蔓延防止、発酵やカビの増殖及び交叉汚染を防止する効果的な措置を講じること。 |
対象医薬品:植物性医薬品 以下の要件があります。 ・検体採取、秤量、混合及び加工作業を行う際に粉塵が生じる場合の製造区域の専用施設の使用について。 |
対象医薬品:治験薬 以下の要件があります。 ・交叉汚染の全リスクを最小化することの必要性が強く求められるため、設備や構造の設計、検査や試験方法及び洗浄後における許容限界についてはこれらリスクの特性を反映すること。 |
対象医薬品:ヒト血液及びヒト血漿由来製品 以下の要件があります。 ・施設と装置は、ウイルス不活性化処理の前と後の製造工程で、分けて使用すること。 |
対象医薬品:原薬 以下の要件があります。 ・ある専用区域から別の専用区域へ移動する従業員、原材料等については、交叉汚染を防止するための適切な対策が必要。 ・工程装置が専用でない場合、交叉汚染を防止するため、異なる原薬等の製造の間に清掃すること。 |
対象医薬品:原薬 以下の要件があります。 ・原材料の受入及び区分保管において、専用ではないタンクによるバルク輸送について。 ・交叉汚染を防止するように、原材料を保管すること。 |
対象医薬品:原薬 以下の要件があります。 ・受託製造業者(試験機関を含む)は、交叉汚染の防止に特別の考慮を払うこと。 ・原薬・中間体の再包装は、交叉汚染を避けるために、適切な環境条件下で実施すること。 |
対象医薬品:原薬(細胞培養・発酵により生産する原薬) 以下の要件があります。 ・多品種製造の共用装置では、交叉汚染のリスクを最小限にするために、キャンペーン製造の間、清掃後の追加の試験が要求されることがある。 |
対象医薬品:生物学的製剤 以下の要件があります。 ・生ワクチンの製造は、他の製品や建物内の他の製造区域への交差汚染を防ぐために、適切な管理のもとで実施されること。 ・専用の製造区域を使用すること。最終容器充填以外のすべての製造工程に対応できる十分なスペースを有すること。 |
対象医薬品:生物学的製剤 以下の要件があります。 ・ワクチン株以外の微生物の存在を伴う可能性のある試験手順や、初代培養以外の組織培養細胞株を使用できない。 |
対象医薬品:生物学的製剤 以下の要件があります。 (ii)複数製品の生産に対応する建物やエリアの場合は交叉汚染防止手順の管理が十分でない限りプロセスの封じ込めが必要。 |
対象医薬品:生物学的製剤 以下の要件があります。 ・異なる生ワクチン製造区域間、または生ワクチン製造区域と他の製造区域との間での製品、設備、及び人員の移動は、交差汚染防止等の条件下で実施すること。 |
対象医薬品:生物学的製剤 以下の要件があります。 ・生ワクチン微生物を適切に除去または除染し、その後に他の製品を製造できるようにするための文書化された手順と効果的なプロセスが整備されること。 |
対象医薬品:生物学的製剤 以下の要件があります。 ・汚染の可能性のある装置や資材は、交叉汚染を防ぐために生産に用いる装置や資材から隔離すること。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 原則と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 製造区域 3.6と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 製造区域 3.8、3.14、3.15と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 保管区域 3.22 品質管理区域 3.27と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第5章 製造 包装作業 5.44と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射線医薬品の製造 建物及び設備 全般事項 17. 製造 34.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 原則 医療用ガスの製造と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 設備 11.と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 保管区域 1.と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 製造区域 5 .と同じ内容となります。 |
対象医薬品:治験薬 別紙(12) PIC/S GMP ガイドライン アネックス13 治験薬の製造 建物及び設備 5. と同じ内容になります。 |
対象医薬品:ヒト血液及びヒト血漿由来製品 PIC/S GMP ガイドライン アネックス14ヒト血液及びヒト血漿由来製品の製造 5. 建物及び設備 5.3 と同じ内容になります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 原則と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 製造区域 3.6と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 製造区域 3.8、3.14、3.15と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 保管区域 3.22 品質管理区域 3.27と同じ内容となります。 |
対象医薬品:一般医薬品 別紙(1) PIC/S GMP ガイドライン パート1 第5章 製造 包装作業 5.44と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射線医薬品の製造 建物及び設備 全般事項 17. 製造 34.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 原則 医療用ガスの製造と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 設備 11.と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 保管区域 1.と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 製造区域 5 .と同じ内容となります。 |
対象医薬品:治験薬 別紙(12) PIC/S GMP ガイドライン アネックス13 治験薬の製造 建物及び設備 5. と同じ内容になります。 |
対象医薬品:ヒト血液及びヒト血漿由来製品 PIC/S GMP ガイドライン アネックス14ヒト血液及びヒト血漿由来製品の製造 5. 建物及び設備 5.3 と同じ内容になります。 |
対象医薬品:全般 以下の要件があります。 ・保管区域において塵埃が発生する場合には、交叉汚染を回避し清掃を行いやすいように適切な措置が取られること。 ・サンプリングが保管区域で行われる場合は、交叉汚染を防止するような方法で実施しなければならない。 ・包装のための建物は交叉汚染を回避できるように設計及び配置しなければならない。 |
対象医薬品:全般 以下の要件があります。 ・建物及び装置は、交叉汚染等製品品質へのいかなる悪影響も回避するための有効な洗浄と保守管理を可能とすること。 ・非専用の装置を用いる場合、異なる製品を製造する前には適切な清掃をおこなうこと。 |
対象医薬品:全般 以下の要件があります。 ・必要に応じエアロックの設置や差圧、給排気システム及び閉鎖系生産システムの採用により交叉汚染を防止すること。 *用語についての補足:「エアロック」は 建築 007:エアロックのWHO 52nd report Annex 8の用語解説を参照ください。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・建築設計における建物において、交叉汚染を防止するよう考慮すること。 ・清浄度や封じ込めの要求仕様の達成と維持が重要である。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | 米国 | ー | ー | 欧州 | 欧州 | ー | ー | ー | ー | ー | ー | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | CDER | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | WHO | WHO | WHO | WHO | ||||||||||||||||||||||||||||||||||
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長 | 厚生労働省医薬食品局審査管理課長 厚生労働省医薬食品局監視指導・麻薬対策課長 |
薬食監麻発 | Code of Federal Regulations | Guidance | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 | 無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
PIC/SのGMPガイドラインを活用する際の考え方について 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
原薬GMPのガイドラインについて | 品質リスクマネジメントに関するガイドライン | 医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン | TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 3 MANUFACTURE OF RADIOPHARMACEUTICALS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 3 Manufacture of Radiopharmaceuticals |
WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS 48th report annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, No. 986, 2014 |
WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS 44th report Annex 2 WHO good manufacturing practices for active pharmaceutical ingredients WHO Technical Report Series, No. 957, 2010 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 55th report Annex 3 Good manufacturing practices: water for pharmaceutical use WHO Technical Report Series, No. 1033, 2021 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 2 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products Part 2: Interpretation of Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1019, 2019 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 3 Good manufacturing practices: guidelines on validation WHO Technical Report Series, No. 1019, 2019 |
||||||||||||||||||||||||||||||||||
| 発行年 | 2021年 | 2011年 | 2020年 | 2012年 | 2021年 | 2006年 | 2012年 | 2023年 | 2004年 | 2021年 | 2023年 | 2015年 | 2008年 | 2014年 | 2010年 | 2021年 | 2018年 | 2019年 | 2019年 | ||||||||||||||||||||||||||||||||||
| 記載内容 | 第二章 医薬品製造業者等の製造所における製造管理及び品質管理 第一節 通則 (製造管理) 第十条 製造業者等は、製造部門に、手順書等に基づき、次に掲げる製造管理に係る業務を適切に行わせなければならない。 八 構造設備を定期的に点検整備するとともに、その記録を作成し、これを保管すること。また、計器の校正を適切に行うとともに、その記録を作成し、これを保管すること。 |
3.品質システム 3.1 品質システム一般要求事項 3) 文書管理 無菌医薬品の無菌性を保証するため、及び本指針各項記載内容に関連する文書として、初期・定期・変更時のバリデーションに関する文書、標準操作手順書(SOP)、清浄度区分図、原材料・職員・中間製品・製品の動線図、機器レイアウト図、各種指図書、記録書、逸脱管理、変更管理及び規格外調査管理、校正記録、環境モニタリング記録、ログブック、コンピューターシステムデータ(電子媒体による記録など)などの文書を作成し、運用と保管をすること。 13. 滅菌工程 13.2.4 日常管理 2) 定期検査、維持管理、校正、装置のテスト項目等を、その具体的な手順及び頻度とともに文書化すること。 |
3.品質システム 3.1 品質システム一般要求事項 3) 文書管理 無菌医薬品の無菌性を保証するため、及び本指針各項記載内容に関連する文書として、初期・定期・変更時のバリデーションに関する文書、標準操作手順書(SOP)、清浄度区分図、原材料・職員・中間製品・製品の動線図、機器レイアウト図、各種指図書、記録書、逸脱管理、変更管理及び規格外調査管理、校正記録、環境モニタリング記録、ログブック、コンピューターシステムデータ(電子媒体による記録など)などの文書を作成し、運用と保管をすること。 13. 滅菌工程 13.2.4 日常管理 2) 定期検査、維持管理、校正、装置のテスト項目等を、その具体的な手順及び頻度とともに文書化すること。 |
12. 製造設備及びユーティリティの適格性評価 12.3 校正 1) 無菌製品に係る製品の無菌性を保証するために重要な制御、測定及びモニタリングに係る各製造設備及びユーティリティの校正のため、責任の割当てその他必要な事項について計画書及び手順書を作成し、これらの文書に従って校正を行うこと。 2) 製造設備及びユーティリティの校正に当たっては、トレーサブル性を確保できる認証された標準器が存在する場合においては、それを用いて実施すること。 3) 上記の校正に係る記録は保管すること。 4) 重要な製造設備及びユーティリティについては、校正に係る現状を認識し、実証することができるようにしておくこと。 5) 校正基準に適合しない計器は使用しないこと。 |
(続き) 6) 無菌製品に係る製品の無菌性を保証するために重要な計器が承認された校正の標準値から逸脱した場合においては、これらの逸脱が、前回の校正以降において当該計器を用いて制御、測定又はモニタリングを行った環境下において製造した無菌製品に係る製品の無菌性に影響を及ぼしたか否かを判定するために、調査及び評価を行うこと。 |
13. 滅菌工程 13.2 高圧蒸気滅菌 13.2.2 滅菌装置 1) 製造者名、型式、寸法、構造、材質、機能、能力等、滅菌装置の主な仕様が文書化されていること。また、通常運転方法のほか、初期設定の方法、異常時の対処方法、分解及び再組立の方法、校正を含む維持管理の方法等が記載された取扱説明書があること。 13.2.3 滅菌バリデーション 10) 温度分布の測定に、装置に装備されている以外の温度計を用いる場合は、試験の前後で校正を行うこと。 |
14. 無菌製造設備の定置清浄化(CIP) 14.4 日常管理 CIP工程毎にそのデータについて記録を作成の上保管し、定期的に照査すること。CIP実施記録その他の記録には、少なくとも以下の事項を記載すること。 9) 洗浄終点を検出する計器、量、圧力等CIPパラメータを示す計器の校正の有効性 15. 無菌製造設備の定置蒸気滅菌(SIP) 15.3 日常管理 5) 温度計等の重要な計器は適切な間隔で校正すること。 |
18. 凍結乾燥工程 18.4 日常管理と維持管理事項 4) 監視及び制御を実施する温度制御器、真空計等の重要計器は定期的に校正し、記録を作成の上保管すること。校正頻度は、前回の結果において頻度変更の必要性が示されない限り、約6カ月毎とすることが望ましい。 5) 真空計は微小圧力を測定する高精度の計器であり、トレーサブル性が確保された方法で現場校正を行うことは現状では困難である。専門の校正機関に依頼し、工場外で校正を実施してもよい。 |
A6.4 外観検査 A6.4.1 一般要件 6) 自動検査機による検査においては,例えば次のような条件を定める ①検査機の機種,検査速度,単位検査対象当たりの所要時間 ② 開始,終了時,その他定期的な標準品サンプル等による検査機の検査能力の確認方法 ③校正 |
第3章 建物及び設備 設備 3.41.測定、秤量、記録及び管理の設備は、所定の間隔で適切な方法により校正し、チェックすること。斯かる試験の適切な記録書を保管すること。 第4章 文書化 製造処方及び工程指図書 4.18 工程指図書は、以下の事項を含むこと。 b) 重要な装置の準備作業( 例えば、清浄化、組立て、校正、滅菌)の方法、又は当該方法の参照先 |
その他 4.29. 以下の例について適宜、文書化された方針、手順書、実施計画書、報告書及び講じた措置又は到達した結論に関連する記録書があること。 − 装置の組立て及び校正 4.31. 主要な又は重要な分析試験、製造装置、及び製品が加工されている区域について、作業記録簿を付けること。作業記録簿は適宜、当該区域の使用、装置/ 方法、校正、保守管理、清浄化又は補修の作業(日付及びそれら作業を行った者の識別を含む) を、時系列に記録するため使用すること。 |
第6 章 品質管理 文書化 6.7 試験施設の文書化は、第4 章に示す原則に従うこと。この文書化の重要部分は品質管理に関するものであり、以下の詳細項目について、品質管理部門が容易に利用可能であること。 (iii) 機器の校正/適格性評価及び設備の保守管理に係る手順及びそれらの記録 |
建物及び設備 20. 予防保全、校正、適格性評価プログラムを行い、放射性医薬品の生産に使用される全ての設備及び装置が適切であり適格とされていることを保証しなければならない。これらは、有能な従業員が行い、記録及び日誌を保管しなければならない。 |
5.工程装置 5.3 校正 5.30 中間体・原薬の品質を保証するために重要な制御、秤量、測定、モニタリング及び試験の各装置については、文書による手順及び計画に従って校正を行うこと。 5.31 装置の校正にあたっては、証明された標準器とのトレーサビリティが確保できる標準器が存在する場合には、これを用いて実施すること。 5.32 上述の校正の記録は保管すること。 5.33 重要な装置については、校正に係る現状を認識し、証明できる状態にしておくこと。 |
5.34 校正基準に適合しない計測器は使用しないこと。 5.35 重要な計測器について承認された校正の標準値から逸脱した場合には、これらの逸脱が前回の校正以降において当該計測器を用いて生産した中間体・原薬の品質に影響を与えたか否かを判定するために、調査を行うこと。 |
付属書Ⅱ :品質リスクマネジメントの潜在用途 Ⅱ .4 施設、設備、ユーティリティのための品質リスクマネジメントbr> 施設/設備/ユーティリティの適格性確認 施設、建物、製造設備及び/又は試験機器に対する適格性確認の範囲と程度を定める(正しいキャリブレーション方法を含む)。 キャリブレーション/予防保全 適切なキャリブレーション及びメンテナンスの日程を決める。 |
別紙-2 カテゴリ3 構成設定していないソフトウェア 商業ベースで販売されている既製のパッケージソフトウェアで、それ自体は業務プロセスに合わせて構成設定していないもの(実行時のパラメータの入力のみで調整されるアプリケーション等は本カテゴリに含まれる) 製造設備、分析機器、製造支援設備等に搭載されるシステム 備考欄 設備に合わせて仕様の設定及び機能の検証を行うことで差し支えない。単純なシステムに関しては校正で代用することも可 |
subpart D-Equipment Sec. 211.68 Automatic, mechanical, and electronic equipment. (a) Automatic, mechanical, or electronic equipment or other types of equipment, including computers, or related systems that will perform a function satisfactorily, may be used in the manufacture, processing, packing, and holding of a drug product. If such equipment is so used, it shall be routinely calibrated, inspected, or checked according to a written program designed to assure proper performance. Written records of those calibration checks and inspections shall be maintained. |
Ⅸ. VALIDATION OF ASEPTIC PROCESSING AND STERILIZATION C. Sterilization of Equipment, Containers, and Closures 1. Qualification and Validation Empty chamber studies evaluate numerous locations throughout a sterilizing unit (e.g., steam autoclave, dry heat oven) or equipment train (e.g., large tanks, immobile piping) to confirm uniformity of conditions (e.g., temperature, pressure). These uniformity or mapping studies should be conducted with calibrated measurement devices. |
(続き) 2. Equipment Controls and Instrument Calibration For both validation and routine process control, the reliability of the data generated by sterilization cycle monitoring devices should be considered to be of the utmost importance. Devices that measure cycle parameters should be routinely calibrated. Written procedures should be established to ensure that these devices are maintained in a calibrated state. For example, we recommend that procedures address the following: ・Temperature and pressure monitoring devices for heat sterilization should be calibrated at suitable intervals. The sensing devices used for validation studies should be calibrated before and after validation runs. |
(続き) ・Devices used to monitor dwell time in the sterilizer should be periodically calibrated. ・Where applicable, instruments used to determine the purity of steam should be calibrated. ・For dry heat depyrogenation tunnels, devices (e.g. sensors and transmitters) used to measure belt speed should be routinely calibrated. |
Ⅺ. STERILITY TESTING 21 CFR 211.160(b) states that “Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedures designed to assure that components, drug product containers, closures, in-process materials, labeling, and drug products conform to appropriate standards of identity, strength, quality, and purity.Laboratory controls shall include: (略)(4) The calibration of instruments, apparatus, gauges, and recording devices at suitable intervals in accordance with an established written program containing specific directions, schedules, limits for accuracy and precision, and provisions for remedial action in the event accuracy and/or precision limits are not met. |
(続き) Instruments, apparatus, gauges, and recording devices not meeting established specifications shall not be used.” |
CHAPTER 3 PREMISES AND EQUIPMENT EQUIPMENT 3.41 Measuring, weighing, recording and control equipment should be calibrated and checked at defined intervals by appropriate methods. Adequate records of such tests should be maintained. CHAPTER 4 DOCUMENTATION MANUFACTURING FORMULA AND PROCESSING INSTRUCTIONS 4.18 The Processing Instructions should include: b) The methods, or reference to the methods, to be used for preparing the critical equipment (e.g. cleaning, assembling, calibrating, sterilising); |
Other 4.29 There should be written policies, procedures, protocols, reports and the associated records of actions taken or conclusions reached, where appropriate, for the following examples: - Equipment assembly and calibration; 4.31. Logbooks should be kept for major or critical analytical testing, production equipment, and areas where product has been processed. They should be used to record in chronological order, as appropriate, any use of the area, equipment/method, calibrations, maintenance, cleaning or repair operations, including the dates and identity of people who carried these operations out. |
CHAPTER 6 Documentation 6.7. Laboratory documentation should follow the principles given in Chapter 4. An important part of this documentation deals with Quality Control and the following details should be readily available to the Quality Control Department: (iii) Procedures for and records of the calibration/qualification of instruments and maintenance of equipment; |
PREMISES AND EQUIPMENT General 20. Preventive maintenance, calibration and qualification programmes should be operated to ensure that all facilities and equipment used in the manufacture of radiopharmaceutical are suitable and qualified. These activities should be carried out by competent personnel and records and logs should be maintained. |
Chapter 3: Premises and Equipment EQUIPMENT 3.41 Measuring, weighing, recording and control equipment should be calibrated and checked at defined intervals by appropriate methods. Adequate records of such tests should be maintained. Chapter 4: Documentation Manufacturing Formula and Processing Instructions 4.18 The Processing Instructions should include: b) The methods, or reference to the methods, to be used for preparing the critical equipment (e.g. cleaning, assembling, calibrating, sterilising); |
Other 4.29 There should be written policies, procedures, protocols, reports and the associated records of actions taken or conclusions reached, where appropriate, for the following examples: - Equipment assembly and calibration; 4.31 Logbooks should be kept for major or critical analytical testing, production equipment, and areas where product has been processed. They should be used to record in chronological order, as appropriate, any use of the area, equipment/method, calibrations, maintenance, cleaning or repair operations, including the dates and identity of people who carried these operations out. |
CHAPTER 6 QUALITY CONTROL Documentation 6.7 Laboratory documentation should follow the principles given in Chapter 4. An important part of this documentation deals with Quality Control and the following details should be readily available to the Quality Control Department: (iii) Procedures for and records of the calibration/qualification of instruments and maintenance of equipment; |
Premises and equipment General 20. Preventive maintenance, calibration and qualification programmes should be operated to ensure that all facilities and equipment used in the manufacture of radiopharmaceutical are suitable and qualified. These activities should be carried out by competent personnel and records and logs should be maintained. |
1. Pharmaceutical quality system 1.5 The PQS appropriate to the manufacture of pharmaceutical products should ensure that: g) all necessary controls on starting materials, intermediate products, and bulk products and other in-process controls, calibrations and validations are carried out; 8. Self-inspection, quality audits and suppliers’ audits and approval Items for self-inspection 8.2 Written instructions for self-inspection should be established to provide a minimum and uniform standard of requirements. These may include questionnaires on GMP requirements covering at least the following items: (k) calibration of instruments or measurement systems; |
13. Equipment 13.5 Balances and other measuring equipment of an appropriate range and precision should be available for production and control operations and should be calibrated according to a fixed schedule. |
15. Documentation Master formulae 15.23 The master formula should include: (f) the methods, or reference to the methods, to be used for preparing and operating the critical equipment, e.g. cleaning (especially after a change in product), assembling, calibrating, sterilizing, use; 15.46 Records should be kept for major and critical equipment, as appropriate, of any validations, calibrations, maintenance, cleaning or repair operations, including dates and the identity of the people who carried out these operations. |
16. Good practices in production Processing operations 16.23 Measuring, weighing, recording, and control equipment and instruments should be serviced and calibrated at prespecified intervals and records maintained. To ensure satisfactory functioning, instruments should be checked daily or prior to use for performing analytical tests. The date of calibration and servicing and the date when recalibration is due should be clearly indicated on a label attached to the instrument. |
5. Process equipment 5.3 Calibration 5.30 Control, weighing, measuring, monitoring and test equipment that is critical for assuring the quality of intermediates or APIs should be calibrated according to written procedures and an established schedule. 5.31 Equipment calibrations should be performed using standards traceable to certified standards, if these exist. 5.32 Records of these calibrations should be maintained. 5.33 The current calibration status of critical equipment should be known and verifi able. |
5.34 Instruments that do not meet calibration criteria should not be used. 5.35 Deviations from approved standards of calibration on critical instruments should be investigated to determine if these could have had an impact on the quality of the intermediate(s) or API(s) manufactured using this equipment since the last successful calibration. |
11. Safe change filter housings 11.4 All filter banks should be provided with pressure differential indication gauges to indicate the filter dust loading and remaining lifespan of the filters. Connection to these gauges should be copper or stainless steel and not plastic tubing, which could perish causing a contamination hazard. The tube connections on the filter casing should be provided with stopcocks, for safe removal or calibration of gauges. |
13. Maintenance of water systems 13.2 The maintenance programme should take into account at least the following: (略); ■ the calibration programme; 14. System reviews 14.3 Examples of matters to be included in the review are :(略); ■ maintenance and calibration history; |
7. Air filtration, airflow direction and pressure differentials 7.8 The pressure control and monitoring devices used should be calibrated. Compliance with specifications should be regularly verified and the results recourded. |
7.Air filtration, airflow direction and pressure differentials
(略) The pressure control and monitoring devices used should be calibrated and, where possible, be linked to an alarm system set according to the determined levels. |
6. Documentation 6.1 Documents associated with qualification and validation may include: (略) ・calibration procedures and records; |
8. Qualification and validation protocols 8.2 As a minimum, the protocols should be appropriate for the qualification or validation to be executed, and may include the following significant background information: (略) ・calibration requirements; |
10. Qualification 10.2 All relevant SOPs for operation, maintenance and calibration should be prepared during qualification. |
(続き) Installation qualification (略) 10.18 Applicable control and measuring devices, identified through impact or risk assessment, should be calibrated. |
(続き) Requalification (略) 10.25 Requalification should be done based on the identified need and risk management principles. Factors such as the frequency of use, breakdowns, results of operation, criticality, preventive maintenance, repairs, calibration, and verification may be considered. |
15. Calibration and verification 15.1 Calibration and verification of equipment, instruments and other devices, as applicable, should be initiated during installation qualification, to ensure that the system operates according to the described specifications and because the calibration status could have been affected during transport and installation. 15.2 Thereafter, it should be performed at regular intervals in accordance with a calibration programme and SOPs. 15.3 Personnel who carry out calibration and preventive maintenance should have appropriate qualification and training. |
15.4 A calibration programme should be available and should provide information such as calibration standards and limits, responsible persons, calibration intervals, records and actions to be taken when problems are identified. 15.5 There should be traceability to standards (e.g. national, regional or international standards) used in the calibration. A valid certificate of calibrationshould be maintained, which is dated and includes reference to and traceability to, for example, standards used, acceptance limits, uncertainty where applicable, range, calibration due date. |
15.6 Calibrated equipment, instruments and other devices should be labelled, coded or otherwise identified, to indicate the status of calibration and the date on which recalibration is due. 15.7 When the equipment, instruments and other devices have not been used for a certain period of time, their function and calibration status should be verified and shown to be satisfactory before use. 15.8 Equipment, instruments and other devices should be calibrated before or on the due date for calibration, to ensure that they are used in a calibrated state. |
15.9 Where instruments and devices are identified as critical or non-critical, or impacting and non-impacting for the purpose of calibration, documented evidence of the decision-making process should be available. This should include impact and/or risk assessment. | Appendix 6 Guidelines on qualification 8. Installation qualification (略) 8.4 Identified measuring, control and indicating devices, should be calibrated on site, unless otherwise appropriately justified. The calibration should be traceable to national or international standards. |
Appendix 6 Guidelines on qualification 9. Operational qualification 9.1 Requirements and procedures for operation (or use), calibration, maintenance and cleaning should be prepared before OQ and approved prior to PQ. (略) 9.5 Calibration, cleaning, maintenance, training and related tests and results should be verified to be acceptable 11. Periodic review and requalification 11.5 Principles of risk management may include factors such as calibration, verification, maintenance data and other information. |
Appendix 7 Non sterile process validation 7. Change management (略) Validation should be considered when changes to production and/or control procedures are planned. Based on risk assessment, changes that may require revalidation could include (but are not limited to): (略) ・changes associated with equipment calibrations and the preventive maintenance carried out, which may impact the process; |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・製造管理者は製造管理に関わる業務として計器の校正とその記録を製造部門に行わせること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌性保証のための文書管理対象のひとつとして校正記録の作成、保管をすること。 ・滅菌工程における日常管理として校正等の項目を、具体的な手順及び頻度とともに文書化すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・製造工程での測定機器、検査機や計測制御デバイスなどは、周期や精度を定めた校正プログラムを構築し、実施すること。 ・浮遊微粒子の測定装置及び浮遊微生物の採取装置について、バリデートされた校正済装置を使用すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・製造設備及びユーティリティの適格性評価において、無菌性を保証するために重要な制御、測定及びモニタリングに係る各製造設備及びユーティリティの校正のため、責任の割当てその他必要な事項について計画書及び手順書を作成し、これらの文書に従って校正を行うこと。 ・その校正においてはトレーサブル性を確保できる認証された標準器が存在する場合においては、それを用いて実施すること。また校正に係る記録は保管すること。 ・重要な製造設備及びユーティリティについては、校正に係る現状を認識し、実証することができるようにしておくこと、またその際に校正基準に適合しない計器は使用しないこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・製品の無菌性を保証するために重要な計器について、承認された校正の標準値から逸脱した場合においては、これらの逸脱が、前回の校正以降において当該計器を用いて制御、測定又はモニタリングを行った環境下において製造した無菌製品に係る製品の無菌性に影響を及ぼしたか否かを判定するために調査及び評価を行うこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・高圧蒸気滅菌装置について、校正を含む維持管理の方法等が記載された取扱説明書があること。 ・滅菌工程の温度分布測定において装置に装備されている以外の温度計を用いる場合は、試験の前後で校正を行うこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CIP工程毎に作成する実施記録には、洗浄終点を検出する計器、流量、圧力等CIPパラメータを示す計器の校正の有効性を記載すること。 ・定置蒸気滅菌(SIP)における温度計等の重要な計器は適切な間隔で校正すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・凍結乾燥工程において 監視及び制御を実施する温度制御器、真空計等の重要計器は定期的に校正し、記録を作成の上保管すること。校正頻度は、前回の結果において頻度変更の必要性が示されない限り、約6カ月毎とすることが望ましい。 ・真空計はトレーサブル性が確保された方法で現場校正を行うことは現状では困難であるため、専門の校正機関に依頼し、工場外で校正を実施してもよい。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・自動検査機による検査においては校正等の条件を定めること。 |
対象医薬品:全般 以下の要件があります。 ・測定、秤量、記録及び管理の設備は、適切な方法によって規定された間隔で校正し、チェックすること。 ・文書の作成と管理において工程指図書は、重要な装置の準備の作業方法、或いは作業方法の参照先(例えば、清掃、組立て、校正、滅菌)を含むこと。 |
対象医薬品:全般 以下の要件があります。 ・必要な場合には、装置の組立て及び校正について、方針、手順、実施計画、報告、行った措置に関連する記録、或いは結論を文書化すること。 ・主要、或いは重要な分析試験、製造装置、製品が製造されている区域の使用記録を保存すること。それらは時系列に、区域、装置/方法、校正、保守、清掃、修理作業を記録するために使用すること。必要に応じて、日付、及びこれらの操作を行う人の識別を含める。 |
対象医薬品:全般 以下の要件があります。 ・試験施設について、機器の校正詳細項目は、品質管理部門が容易に利用可能であること。 |
対象医薬品:放射性医薬品 以下の要件があります。 ・校正等について、生産に使用される全ての設備及び装置が適切であり適格とされていることを保証すること。 ・同作業は有能な従業員が行い、記録及び日誌を保管すること。 |
対象医薬品:原薬 以下の要件があります。 ・中間体・原薬の品質を保証するために重要な制御、秤量、測定、モニタリング及び試験の各装置について、文書による手順及び計画に従って校正を行うこと。 ・装置の校正にあたっては、証明された標準器とのトレーサビリティが確保できる標準器が存在する場合には、これを用いて実施すること、また校正の記録は保管すること。 ・ 重要な装置については、校正に係る現状を認識し、証明できる状態にしておくこと。 |
対象医薬品:原薬 以下の要件があります。 ・校正基準に適合しない計測器は使用しないこと。 ・重要な計測器について承認された校正の標準値から逸脱した場合には、これらの逸脱が前回の校正以降において当該計測器を用いて生産した中間体・原薬の品質に影響を与えたか否かを判定するために、調査を行うこと。 |
対象医薬品:全般 以下の要件があります。 ・施設、建物、製造設備についての適格性確認として正しいキャリブレーション方法を含めて定めること。 ・適切なキャリブレーション日程を定めること。 |
対象医薬品:全般 以下の要件があります。 ・カテゴリ3 構成設定していないソフトウェアのうち製造設備、分析機器、製造支援設備等に搭載されるシステムについて、単純なシステムに関しては校正で代用することも可。 |
対象医薬品:全般 以下の要件があります。 ・自動式、機械式、電子式装置(コンピュータ等を含む)を加工、包装、保管に用いるとき、適正に稼動していることを日常的に校正し、検査または確認すること。 ・校正確認や検査の記録書を保管すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・滅菌ユニットや装置ライン内が均一な環境(温度、圧力等)であることを評価しなければならないが、その均一性や分布調査には校正された装置を用いること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・滅菌サイクルにおけるパラメータを計測する機器は日常的に校正されている必要がある。 ・その校正の管理は文書化されていること。 ・例えば加熱滅菌装置における温度と圧力モニタリングセンサーについては適切に校正されること。またバリデーションに用いるセンサーの場合、バリデーションの前後で校正を行うこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・滅菌機における滞留時間を検出する装置は、定期的に校正を行うこと。 ・蒸気の純度を計測する装置は、校正を行うこと。 ・乾熱滅菌トンネルのベルトスピードに用いられるセンサーや伝達装置は、日常的に校正を行うこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌試験について。試験室管理として、装置、機械、計器、記録計の校正を行うこと。 ・プロクラムに合致した適切な期間の校正を行うこと。 ・プロクラムには、具体的な手順やスケジュール、正確度と精度の限度、正確度、精度が達成されなかった場合の規定された改善措置を含むこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌試験について。試験室管理として、設定した基準を逸脱したの装置、機械、計器、記録計は使用しないこと。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 設備 3.41.と同じ内容となります。 第4章 文書化 製造処方及び工程指図書 4.18.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第4章 文書化 その他 4.29.、4.31.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第6章 品質管理 文書化 6.7. と同じ内容となります。 |
対象医薬品:放射性医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 建物及び設備 20.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 設備 3.41.と同じ内容となります。 第4章 文書化 製造処方及び工程指図書 4.18.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第4章 文書化 その他 4.29.、4.31.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第6章 品質管理 文書化 6.7. と同じ内容となります。 |
対象医薬品:放射性医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 建物及び設備 20.と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・出発物質、中間製品、バルク製品及び他の工程内管理に対し、校正を実施すること。 ・自己点検について。指示書の質問事項には、GMP要件として装置や計測システムの校正が行われていることを含むこと。 |
対象医薬品:全般 以下の要件があります。 ・天秤等の装置は定められた期間で校正すること。 |
対象医薬品:全般 以下の要件があります。 ・標準処方には準備や操作に用いる重要機器の手法、例えば校正等が含まれること。 ・バリデーション、校正、保守、清掃あるいは修理について全ての記録を保管すること。記録には日付と実施者名を含むこと。 |
対象医薬品:全般 以下の要件があります。 ・測定、秤量、記録及び管理の設備は、適切な方法によって規定された間隔で校正し、チェックすること。 ・校正実施日と次回校正日を明記したラベルを装置につけること。 |
対象医薬品:原薬 原薬GMPのガイドラインについて 5.工程装置 5.3 校正 5.30、5.31、5.32、5.33と同じ内容となります。 |
対象医薬品:原薬 原薬GMPのガイドラインについて 5.工程装置 5.3 校正 5.34、5.35と同じ内容となります。 |
対象医薬品:ハザード物質 以下の要件があります。 ・排気フィルタ安全交換型装置において、フィルタ交換時期を確認するための差圧計は、安全に取り外しや校正が行えるよう閉止弁を介してチューブ接続すること。 |
対象医薬品:製薬用水 以下の要件があります。 ・保守プログラムには、校正プログラムを考慮すること。 ・システムレビューにおいては、校正と保守の履歴を含むこと。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・使用される差圧制御装置および差圧モニタリング装置は、校正を行うこと。また仕様どおりであることを定期的に検証し、記録として残すこと。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・差圧制御装置および差圧モニタリング装置は、校正を行い評価すること、また可能であれば所定のレベルにおける警報システムとリンクさせること。 |
対象医薬品:記述なし 以下の要件があります。 ・適格性評価とバリデーションのドキュメントに、校正手順および記録を含むこと。 |
対象医薬品:記述なし 以下の要件があります。 ・適格性評価とバリデーションのプロトコルには、重要な情報として校正要件の情報を含むこと。 |
対象医薬品:記述なし 以下の要件があります。 ・操作、メンテナンス、および校正に関連するすべての SOP は、認定時に準備する必要があること。 |
対象医薬品:記述なし 以下の要件があります。 インパクトあるいはリスクアセスメントで特定された該当する制御装置と測定装置は校正されるべきであること。 |
対象医薬品:記述なし 以下の要件があります。 再評価は特定されたニーズとリスクマネージメントの原則に基づくこと。 使用頻度、故障、運転結果、重要度、予防保全、修理、校正およびバリデーションなどの要因が考慮される場合があること。 |
対象医薬品:記述なし 以下の要件があります。 ・校正は、輸送および設置の過程で校正状態が影響を受けている可能性があるためIQ段階で実施すること。 ・校正プログラムおよびSOPに従って、定期的な間隔で実施すること。 ・校正を行う要員は、適切な資格と訓練を受けていること。 |
対象医薬品:記述なし 以下の要件があります。 ・校正プログラムは整備されていること。プロクラムには、校正基準および許容範囲、責任者、校正の間隔、記録、ならびに問題が特定された場合の措置を含むこと。 ・トレーサビリティが確保されていること。 ・校正証明書は有効であること。 ・校正証明書は、日付、使用された基準、許容限界、該当する場合の不確かさ、測定範囲、次回校正予定日を参照できること。 |
対象医薬品:記述なし 以下の要件があります。 ・校正された装置、計器、デバイスに対し、校正の状態および再校正の予定日を示すために、ラベル付け、コード化すること。 ・一定期間使用されていなかった装置、計器、デバイスに対し、には、使用前に校正状態を確認すること。 ・装置、計器、デバイスに対し、再校正の予定日までに校正し、校正された状態で使用できること。 |
対象医薬品:記述なし 以下の要件があります。 ・装置、計器、デバイスに対し、重要度について、文書化すること。文書には影響評価および/またはリスク評価が含まれていること。 |
対象医薬品:記述なし 以下の要件があります。 ・正当な理由がある場合を除き、現場で校正すること。 ・校正は国内または国際標準に追跡可能なトレーサビリティが確保されていること。 |
対象医薬品:記述なし 以下の要件があります。 ・校正、メンテナンスおよび洗浄の要件と手順はOQ以前に準備し、PQに先立って承認すること。 ・校正が許容できるが検証すること。 ・定期的なレビューと再評価について、リスクマネージメントの原則に校正のデータ、情報などの要因を含むこと。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・リスクアセスメントに基づき、再バリデーションとなる変更がある。機器の校正による変更が含まれる。 |
| 国 | 日本 | 日本 | ー | ー | ー |
|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | WHO | WHO | WHO |
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 厚生労働省医薬局長 | GMP | GMP | GMP |
| 法規類 タイトル |
最終滅菌法による無菌医薬品の製造に関する指針 | 原薬GMPのガイドラインについて | WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS 48th report Annex 2 WHO good manufacturing practices for pharmaceutical products: main principles WHO Technical Report Series, No. 986, 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 44th report Annex 2 WHO good manufacturing practices for active pharmaceutical ingredients WHO Technical Report Series, No. 957, 2010 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 53rd report Annex 3 Good manufacturing practices: guidelines on validation WHO Technical Report Series, No. 1019, 2019 |
| 発行年 | 2012年 | 2001年 | 2014年 | 2010年 | 2019年 |
| 記載内容 | 2. 用語定義又は説明 2.10 校正(calibration):一定の条件下で、機器あるいは測定システム(特に秤量においては)、記録機器、及び制御器により示される値あるいは測定で得られた値とそれに対応する適正な標準器の既知の値との相関を確立する一連の操作をいう、予め測定結果の許容範囲を定めておくこと。 |
20 用語集 校正 特定の計器又は装置が、適切な測定の範囲において、ある対照品又は追跡可能な標準品との比較によって、規定した限界値内の結果を示すことを実証すること。 |
Glossary calibration. The set of operations that establish, under specified conditions, the relationship between values indicated by an instrument or system for measuring (especially weighing), recording, and controlling, or the values represented by a material measure, and the corresponding known values of a reference standard. Limits for acceptance of the results of measuring should be established. |
20. Glossary calibration The demonstration that a particular instrument or device produces results within specifi ed limits by comparison with those produced by a reference or traceable standard over an appropriate range of measurements. |
3. Glossary (略) calibration. The set of operations that establish, under specified conditions, the relationship between values indicated by an instrument or system for measuring (especially weighing), recording, and controlling, or the values represented by a material measure, and the corresponding known values of a reference standard. Limits for acceptance of the results of measuring should be established. |
| 概 要 | 対象医薬品:最終滅菌法による無菌医薬品 以下の要件があります。 ・校正の定義 記録機器,及び制御器により示される値あるいは測定で得られた値と、適正な標準器の既知の値との相関を確立する一連の操作のこと。 ・特に秤量において必要である。 ・予め測定結果の許容範囲を定めておくこと。 |
対象医薬品:原薬 以下の要件があります。 ・校正の定義 ある対照品又は追跡可能な標準品との比較によって、規定した限界値内の結果を示すことを実証すること。 |
対象医薬品:全般 最終滅菌法による無菌医薬品の製造に関する指針 2. 用語定義又は説明 2.10 と同じ内容となります。 |
対象医薬品:原薬 原薬GMPのガイドラインについて 20 用語集と同じ内容となります |
対象医薬品:全般 最終滅菌法による無菌医薬品の製造に関する指針 2. 用語定義又は説明 2.10 と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | ー | ー | ー | ー | ー | ー | ー | 欧州 | 欧州 | 欧州 | 欧州 | 欧州 | 欧州 | ー | ー | ー | |||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | PIC/S | PIC/S | PIC/S | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | WHO | |||||||||||||||||||||||||||||
| 分 類 | 省令 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||||||||||||||||||||||||||
| 法規類 タイトル |
医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 | 無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1) PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙 (3)PIC/S GMPガイドライン アネックス2B ヒト用生物学的医薬品( 原薬及び製剤) の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(14) PIC/S GMP ガイドライン アネックス15 クオリフィケーション及びバリデーション |
「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(15) PIC/S GMP ガイドライン アネックス17 パラメトリックリリース |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 2B MANUFACTURE OF BIOLOGICAL MEDICINAL SUBSTANCES AND PRODUCTS FOR HUMAN USE |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 3 MANUFACTURE OF RADIOPHARMACEUTICALS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 6 MANUFACTURE OF MEDICINAL GASES |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 15 QUALIFICATION AND VALIDATION |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 17 REAL TIME RELEASE TESTING AND PARAMETRIC RELEASE |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Part I - Basic Requirements for Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 3 Manufacture of Radiopharmaceuticals |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 6 Manufacture of Medicinal Gases |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 15 Qualification and Validation |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 17: Real Time Release Testing and Parametric Release |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
48th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products: main principles WHO Technical Report Series, No. 986, 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010, 2018 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||||||||||||||||||||||||||||
| 発行年 | 2021年 | 2011年 | 2020年 | 2025年 | 2022年 | 2012年 | 2013年 | 2015年 | 2020年 | 2021年 | 2023年 | 2023年 | 2023年 | 2023年 | 2023年 | 2023年 | 2015年 | 2023年 | 2008年 | 2010年 | 2015年 | 2018年 | 2014年 | 2018年 | 2022年 | |||||||||||||||||||||||||||||
| 記載内容 | 第二章 医薬品製造業者等の製造所における製造管理及び品質管理 第一節 通則(構造設備) 第九条 製品の製造所の構造設備は、次に定めるところに適合するものでなければならない。 一 手順書等に基づき、その用途に応じ適切に清掃及び保守が行われ、必要に応じ殺菌され、また、その記録が作成され、保管されていること。 (製造管理) 第十条 製造業者等は、製造部門に、手順書等に基づき、次に掲げる製造管理に係る業務を適切に行わせなければならない。 九 構造設備を定期的に点検整備するとともに、その記録を作成し、これを保管すること。また、計器の校正を適切に行うとともに、その記録を作成し、これを保管すること。 |
4.職員 4.1 職員の教育訓練 3)無菌操作法に係る作業に関する教育訓練には、少なくとも以下の事項が含まれるものとすること。これらの内容を全て同時に行う必要はないが、文書化された計画に基づいて逐次実施すること。その内容及び実施頻度は、作業の内容並びに職員の知識・技能及び経験に応じて定められるものであること。 ④ 更衣手順 ・ 無菌操作区域の製造休止時に無菌状態を解除し、設備等の点検又は保全のために入室する場合においても、その服装と手順について教育訓練を行うこと。その教育訓練には持込機材の取扱いを含むこと。当該教育訓練を受けていない者(設備業者を含む。)が入室する場合においては、教育を受けたものが入室時に立会い、更衣及び持込機材の取扱いについて説明を行うこと。 |
第2章 人員 主要責任者 2.7 製造部門の長は一般的に、以下の責務を有する。 (iv) 自らの部門、建物及び設備の適格性評価及び保守管理を確保する。 2.8 品質管理部門の長は一般的に、以下の責務を有する。 (v) 自らの部門、建物及び設備について、適格性評価及び保守管理を確保する。 |
教育訓練 2.10 製造業者は、職責により製造及び貯蔵区域又は管理試験施設に立ち入る人員全て(技術、保守管理及び清浄化の人員を含む) 並びにその行動が製品品質に影響を及ぼし得る他の人員に、教育訓練を実施すること。 |
第3章 建物及び設備 原則 実施される作業に支障を生じないよう、建物及び設備を配置し、設計し、建造し、構成し、保守管理しなければならない。その配置及び設計は、過誤のリスクを最小化する旨を目途とするとともに、交叉汚染、塵埃又は汚れの蓄積及び( 一般的に) 製品品質への悪影響を回避するために、有効な清浄化及び保守管理を可能とするものでなければならない。 |
(続き) その他 4.29. 以下の例について適宜、文書化された方針、手順書、実施計画書、報告書及び講じた措置又は到達した結論に関連する記録書があること。 - 装置の組立て及び校正 4.31. 主要な又は重要な分析試験、製造装置、及び製品が加工されている区域について、作業記録簿を付けること。作業記録簿は適宜、当該区域の使用、装置/ 方法、校正、保守管理、清浄化又は補修の作業(日付及びそれら作業を行った者の識別を含む) を、時系列に記録するため使用すること。 |
第6 章 品質管理 文書化 6.7 試験施設の文書化は、第4 章に示す原則に従うこと。この文書化の重要部分は品質管理に関するものであり、以下の詳細項目について、品質管理部門が容易に利用可能であること。 (iii) 機器の校正/適格性評価及び設備の保守管理に係る手順及びそれらの記録 |
2 原則 2.5 (略) CCS の中で検討すべき要素には、以下を含めること(ただし、これらに限定されるものではない) 。 ⅻ 予防的な保守管理ー汚染の追加的リスクがないことを確保する水準まで設備、ユーティリティ及び建物を保守管理する( 計画された保守管理及び計画外の保守管理) |
7 人員 7.3 清浄化、保守管理、モニタリングを実行する者及びクリーンルームに立ち入る者を含む全ての人員は、定期的な教育訓練、作業衣着用の適格性評価及び無菌製品の適正製造に関連する専門分野における評価を受けること。この教育訓練には、微生物学及び衛生学の基礎を含めること、また、クリーンルーム実務、汚染制御、無菌操作技術及び無菌製品の保護(グレードB クリーンルームに入室する作業者及び/又はグレードA への介入操作を行う作業者向け) 並びに製品が無菌でないときの患者に対する安全性上の潜在的影響に特に重点が置かれること。教育訓練のレベルは、その人員が従事する役割及び区域の重要度に基づくものであること。 |
9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.11 モニタリング手順には、傾向分析への対応を規定すること。傾向分析には、以下を含めること( ただし、これらに限定されるものではない): iii 処置限度値からの単発的だが定期的に生じる外れ値で、共通した原因を有するおそれがあるもの(例:計画された予防的な保守管理の後で常に生じる単発の外れ値) |
用語解説 汚染制御ストラテジー(CCS) - 微生物、エンドトキシン/発熱性物質及び微粒子を制御する計画一式であり、現行の製品及び工程の理解から導き出され、工程性能及び製品品質を保証するもの。当該制御には、原薬、添加剤並びに薬剤の原材料及び構成物、施設及び設備の操作条件、工程内管理、最終製品の規格、並びに付随するモニタリング及び管理の頻度及び方法に関連するパラメータ及び特性が含まれ得る。 |
パートA. 一般的ガイダンス 人員 1. 生物学的原薬及び製剤を製造し、試験する区域内で従事する人員(清浄化、保守管理又は品質管理の関係者を含む)は、製造する製品及びその作業(製品、人員及び環境を保護する特定のセキュリティ措置を含む) に特化した教育訓練及び定期的な再教育訓練を受けること。 2. 製品安全のために、人員の健康状態を考慮に入れること。(必要な場合)製造、保守管理、試験並びに使用動物の世話(及び検査) に従事する人員は、適切なワクチン接種及び定期的な健康診断を受けること。 |
人員 14. 放射性製品を生産する区域で働く全ての従業員(清掃及び設備保全に関与する従業員を含む)は、このクラスの製品に適応した追加の教育訓練を受けなければならない。 文書化 32. 主要な装置の使用、清掃、消毒・滅菌及び保守に係る記録には、日付、時間、これらの活動を行った担当者の署名に加えて、該当する場合、製品名及びロット番号を記載しなければならない。 |
職員 6.(シリンダー、或いはバルブの保守点検を担当する職員のような)医療用ガスの品質に影響を及ぼしうる受託業者の職員は適切な教育訓練を受けること。 シリンダーと移動型極低温容器への充填と表示 28.シリンダー、移動型極低温容器及びバルブの保守管理及び修理作業は、医薬品の製造業者の責任である。それらの作業を業者に委託する場合には、受託業者の承認及び技術協定を含む契約締結をすること。受託業者が実施する作業が適切な基準を維持していることを確認するため、受託業者の監査を実施すること。 |
3.設備、施設、ユーティリティ及びシステムのクオリフィケーション段階 運転時適格性評価 (OQ) 3.12 OQが成功裡に完了することにより、作業標準及び洗浄手順、作業者のトレーニング、及び予防的メンテナンスの要求事項を完成することが出来るはずである。 11. 変更管理 11.4 計画された変更について、製品品質、医薬品質システム、文書化、バリデーション、薬事上の現状、キャリブレーション、メンテナンス、及び他のいかなるシステムにおいても、予期しない結果を避け、必要なプロセスバリデーション、ベリフィケーションあるいは再適格性評価等の業務を計画するために品質リスク管理を用いること。 |
4. パラメトリックリリース及び滅菌工程 4.5 無菌性保証プログラムを文書化するとともに、少なくとも、重要工程パラメータの特定及びモニタリング、滅菌サイクルの開発及びバリデーション、容器/ 包装の完全性バリデーション、バイオバーデン管理、環境モニタリングプログラム、製品の隔離保管計画、設備、付帯設備及び施設の設計及び適格性評価プログラム、保守管理及び校正プログラム、変更管理プログラム、人員の教育訓練、並びに品質リスクマネジメント対応の取入れを含めること。 |
滅菌工程 4.18 パラメトリックリリースが規制当局 によって承認されている場合には、バッチの出荷可否判定は、当該承認規格及び重要工程管理データの照査に基づくこと。滅菌器の日常点検、変更、逸脱、不定期及び定期の保守管理活動を記録し、評価し、市場への製品出荷前に承認すること。(* 訳注) 最終製品が無菌試験に合格していても、パラメトリックリリースに係る規格の不適合判定を覆すことはできない。 * 訳注: 日本では、当該製品の製造販売業者が市場への出荷可否の決定に際して評価する責任を有する。 |
CHAPTER 2 PERSONNEL KEY PERSONNEL 2.7 The head of Production generally has the following responsibilities: (iv) To ensure the qualification and maintenance of his department, premises and equipment; 2.8 The head of Quality Control generally has the following responsibilities: (v) To ensure the qualification and maintenance of his/her department, premises and equipment; |
TRAINING 2.10 The manufacturer should provide training for all the personnel whose duties take them into production and storage areas or into control laboratories (including the technical, maintenance and cleaning personnel), and for other personnel whose activities could affect the quality of the product |
CHAPTER 3 PREMISES AND EQUIPMENT PRINCIPLE Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt and, in general, any adverse effect on the quality of products. |
(続き) Other 4.29 There should be written policies, procedures, protocols, reports and the associated records of actions taken or conclusions reached, where appropriate, for the following examples: - Equipment assembly and calibration; 4.31. Logbooks should be kept for major or critical analytical testing, production equipment, and areas where product has been processed. They should be used to record in chronological order, as appropriate, any use of the area, equipment/method, calibrations, maintenance, cleaning or repair operations, including the dates and identity of people who carried these operations out. |
CHAPTER 6 Documentation 6.7. Laboratory documentation should follow the principles given in Chapter 4. An important part of this documentation deals with Quality Control and the following details should be readily available to the Quality Control Department: (iii) Procedures for and records of the calibration/qualification of instruments and maintenance of equipment; |
2 Principle 2.5 (略) Elements to be considered within a CCS should include (but are not limited to): xii. preventative maintenance - maintaining equipment, utilities and premises (planned and unplanned maintenance) to a standard that will ensure there is no additional risk of contamination; |
7 Personnel 7.3 All personnel including those performing cleaning, maintenance, monitoring and those that access cleanrooms should receive regular training, gowning qualification and assessment in disciplines relevant to the correct manufacture of sterile products. This training should include the basic elements of microbiology and hygiene, with a specific focus on cleanroom practices, contamination control, aseptic techniques and the protection of sterile products (for those operators entering the grade B cleanrooms and/or intervening into grade A) and the potential safety implications to the patient if the product is not sterile. The level of training should be based on the criticality of the function and area in which the personnel are working. |
9 Environmental & process monitoring ENVIRONMENTAL AND PROCESS MONITORING 9.11Monitoring procedures should define the approach to trending. Trends should include, but are not limited to: iii. regular but isolated excursion from action limits that may have a common cause, (e.g. single excursions that always follow planned preventative maintenance) |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
PART A: GENERAL GUIDANCE OPERATING PRINCIPLES PERSONNEL 1. Personnel (including those concerned with cleaning, maintenance or quality control) employed in areas where biological active substances and products are manufactured and tested should receive training, and periodic retraining, specific to the products manufactured and to their work, including any specific security measures to protect product, personnel and the environment. 2. The health status of personnel should be taken into consideration for product safety. Where necessary, personnel engaged in production, maintenance, testing and animal care (and inspections) should be vaccinated with appropriate specific vaccines and have regular health checks. |
PERSONNEL 14. All personnel (including those concerned with cleaning and maintenance) employed in areas where radioactive products are manufactured should receive additional training adapted to this class of products. DOCUMENTATION 32. Records of major equipment use, cleaning, sanitisation or sterilisation and maintenance should show the product name and batch number, where appropriate, in addition to the date and time and signature for the persons involved in these activities. |
PERSONNEL 6. Personnel of subcontractors that could influence the quality of medicinal gases (such as personnel in charge of maintenance of cylinders or valves) should be appropriately trained. Filling and labelling of cylinders and mobile cryogenic vessels 28. Maintenance and repair operations of cylinders, mobile cryogenic vessels and valves are the responsibility of the manufacturer of the medicinal product. If subcontracted, they should only be carried out by approved subcontractors, and contracts including technical agreements should be established. Subcontractors should be audited to ensure that appropriate standards are maintained. |
3.QUALIFICATION STAGES FOR EQUIPMENT, FACILITIES, UTILITIES AND SYSTEMS. Operational qualification (OQ) 3.12 The completion of a successful OQ should allow the finalisation of standard operating and cleaning procedures, operator training and preventative maintenance requirements. 11. CHANGE CONTROL 11.4 Quality risk management should be used to evaluate planned changes to determine the potential impact on product quality, pharmaceutical quality systems, documentation, validation, regulatory status, calibration, maintenance and on any other system to avoid unintended consequences and to plan for any necessary process validation, verification or requalification efforts. |
4. PARAMETRIC RELEASE AND STERILISATION 4.5 The sterility assurance program should be documented and include, at least, the identification and monitoring of the critical process parameters, steriliser cycle development and validation, container/packaging integrity validation, bioburden control, environmental monitoring program, product segregation plan, equipment, services and facility design and qualification program, maintenance and calibration program, change control program, personnel training, and incorporate a quality risk management approach. |
Sterilisation Process 4.18 Once parametric release has been approved by the regulatory authorities, decisions for release or rejection of a batch should be based on the approved specifications and the review of critical process control data. Routine checks of the steriliser, changes, deviations, unplanned and routine planned maintenance activities should be recorded, assessed and approved before releasing the products to the market. Non-compliance with the specification for parametric release cannot be overruled by a finished product passing the test for sterility. |
Chapter 2: Personnel Key Personnel 2.7 The head of the Production Department generally has the following responsibilities: iv. To ensure the qualification and maintenance of his department, premises and equipment; 2.8 The head of Quality Control generally has the following responsibilities: v. To ensure the qualification and maintenance of his department, premises and equipment; Training |
2.10 The manufacturer should provide training for all the personnel whose duties take them into production and storage areas or into control laboratories (including the technical, maintenance and cleaning personnel), and for other personnel whose activities could affect the quality of the product. | Chapter 3: Premises and Equipment PRINCIPLE Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt and, in general, any adverse effect on the quality of products. |
Chapter 4: Documentation Other 4.29 There should be written policies, procedures, protocols, reports and the associated records of actions taken or conclusions reached, where appropriate, for the following examples: ー Maintenance, cleaning and sanitation; 4.31 Logbooks should be kept for major or critical analytical testing, production equipment, and areas where product has been processed. They should be used to record in chronological order, as appropriate, any use of the area, equipment/method, calibrations, maintenance, cleaning or repair operations, including the dates and identity of people who carried these operations out. |
Chapter 6: Quality Control Documentation 6.7 Laboratory documentation should follow the principles given in Chapter 4. An important part of this documentation deals with Quality Control and the following details should be readily available to the Quality Control Department: iii. Procedures for and records of the calibration/qualification of instruments and maintenance of equipment; |
2 Principle 2.5 (略) Elements to be considered within a CCS should include (but are not limited to): xii. Preventative maintenance - maintaining equipment, utilities and premises (planned and unplanned maintenance) to a standard that will ensure there is no additional risk of contamination. |
7 Personnel 7.3 All personnel including those performing cleaning, maintenance, monitoring and those that access cleanrooms should receive regular training, gowning qualification and assessment in disciplines relevant to the correct manufacture of sterile products. This training should include the basic elements of microbiology and hygiene, with a specific focus on cleanroom practices, contamination control, aseptic techniques and the protection of sterile products (for those operators entering the grade B cleanrooms and/or intervening into grade A) and the potential safety implications to the patient if the product is not sterile. The level of training should be based on the criticality of the function and area in which the personnel are working. |
9 Environmental & process monitoring Environmental and process monitoring 9.11 Monitoring procedures should define the approach to trending. Trends should include, but are not limited to: iii. Regular but isolated excursion from action limits that may have a common cause, (e.g. single excursions that always follow planned preventative maintenance). |
Glossary Contamination Control Strategy (CCS) - A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
Personnel 14. All personnel (including those concerned with cleaning and maintenance) employed in areas where radioactive products are manufactured should receive appropriate additional training specific to these types of procedures and products Documentation 32. Records of major equipment use, cleaning, sanitisation or sterilisation and maintenance should show the product name and batch number, where appropriate, in addition to the date and time and signature for the persons involved in these activities. |
Personnel 6. Personnel of subcontractors that could influence the quality of medicinal gases (such as personnel in charge of maintenance of cylinders or valves) should be appropriately trained. Filling and labelling of cylinders and mobile cryogenic vessels 28. Maintenance and repair operations of cylinders, mobile cryogenic vessels and valves are the responsibility of the manufacturer of the medicinal product. If subcontracted, they should only be carried out by approved subcontractors, and contracts including technical agreements should be established. Subcontractors should be audited to ensure that appropriate standards are maintained. |
3. QUALIFICATION STAGES FOR EQUIPMENT, FACILITIES, UTILITIES AND SYSTEMS. Operational qualification (OQ) 3.12. The completion of a successful OQ should allow the finalisation of standard operating and cleaning procedures, operator training and preventative maintenance requirements.; 11. CHANGE CONTROL 11.4. Quality risk management should be used to evaluate planned changes to determine the potential impact on product quality, pharmaceutical quality systems, documentation, validation, regulatory status, calibration, maintenance and on any other system to avoid unintended consequences and to plan for any necessary process validation, verification or requalification efforts. |
4. Parametric release and sterilization 4.5 The sterility assurance program should be documented and include, at least, the identification and monitoring of the critical process parameters, sterilizer cycle development and validation, container/packaging integrity validation, bioburden control, environmental monitoring program, product segregation plan, equipment, services and facility design and qualification program, maintenance and calibration program, change control program, personnel training, and incorporate a quality risk management approach. |
Sterilization Process 4.18 Once parametric release has been approved by the regulatory authorities, decisions for release or rejection of a batch should be based on the approved specifications and the review of critical process control data. Routine checks of the sterilizer, changes, deviations, unplanned and routine planned maintenance activities should be recorded, assessed and approved before releasing the products to the market. Non-compliance with the specification for parametric release cannot be overruled by a finished product passing the test for sterility. |
9. Personnel key personnel 9.9 The head of the production generally has the following responsibilities: (略) (d) to check the maintenance of the department, premises and equipment; (略) |
10. Training 10.1 The manufacturer should provide training in accordance with a written programme for all personnel whose duties take them into manufacturing areas or into control laboratories (including the technical, maintenance and cleaning personnel) and for other personnel as required. |
15. Documentation 15.31 SOPs and associated records of actions taken or, where appropriate, conclusions reached should be available for: (略) (c) maintenance, cleaning and sanitization; (略) 15.46 Records should be kept for major and critical equipment, as appropriate, of any validations, calibrations, maintenance, cleaning, or repair operations, including dates and the identity of the people who carried these operations out. |
3. Glossary standard operating procedure. An authorized written procedure, giving instructions for performing operations, not necessarily specific to a given product or material, but of a more general nature (for example operation of equipment, maintenance and cleaning, validation, cleaning of premises and environmental control, sampling and inspection). Certain standard operating procedures may be used to supplement product-specific master and batch production documentation. |
2. Principle 2.5 (略) Elements to be considered within a CCS should include: xii. maintenance of equipment, utilities and premises (planned and unplanned maintenance); |
7. Personnel 7.3 Personnel, including those performing cleaning, maintenance and monitoring and those that access cleanrooms, should receive regular training and undergo gowning qualification and assessment in disciplines relevant to the correct manufacture of sterile products. This training should include the basic elements of microbiology and hygiene (with a specific focus on cleanroom practices), contamination control, aseptic techniques and the protection of sterile products (for those operators entering the grade B cleanrooms or intervening into grade A), and the potential safety implications for the patient if the product is not sterile. The level of training should be based on the criticality of the function and area in which the personnel are working. |
9. Environmental and process monitoring Environmental and process monitoring 9.11 Monitoring procedures should define the approach to trending. Trends should include: iii. regular but isolated excursion from action limits that may have a common cause (for example, single excursions that always follow planned preventive maintenance); |
Glossary contamination control strategy (CCS). A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 製造所の構造設備 ・保守が行われ、また、その記録が作成され、保管されていること。 ・製造業者等は、手順書等に基づき、製造部門に以下の業務を行わせること。構造設備を定期的に点検整備、点検整備の記録を作成、保管。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌状態を解除し、設備等の点検又は保全のために入室する場合において、その服装と手順について教育訓練を行うこと。 |
対象医薬品:全般 以下の要件があります。 ・製造部門の長は自らの部門、建物及び設備の保守管理を保証すること。 ・品質管理部門の長は自らの部門、建物及び設備の保守管理を保証すること。 |
対象医薬品:全般 以下の要件があります。 ・製造業者は、職責により製造区域及び保管区域又は管理試験室に立ち入る保守管理を含む全ての人員に教育訓練を実施すること。 |
対象医薬品:全般 以下の要件があります。 ・建物及び設備は、保守管理すること。配置及び設計は、保守管理を可能とすること。 |
対象医薬品:全般 以下の要件があります。 ・保守管理について文書化された方針、手順書、実施計画書、報告書、講じられた措置に関連する記録書、又は結論書があること。 ・重要な分析試験、製造装置、及び製品が加工されている区域について、保守管理時には作業記録簿(時系列に記録するため使用する)を付けること。 |
対象医薬品:全般 以下の要件があります。 ・試験室の保守管理に関する手順及び記録の詳細な項目についての文書は速やかに品質管理部門において利用可能であること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCS の中で検討すべき要素に、予防的な保守管理について含めること。汚染の追加的リスクがないことを確保する水準まで設備、ユーティリティ及び建物を保守管理すること。 *用語ついての補足:「CCS」は本表アネックス1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・保守管理の人員は、定期的な教育訓練、作業衣着用の適格性評価及び無菌製品の適正製造に関連する専門分野における評価を受けること。教育訓練には、微生物学及び衛生学の基礎を含めること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・モニタリング手順で規定する傾向分析への対応について。傾向分析には、計画された予防的な保守管理の後で常に生じる単発の外れ値を含めること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・CCSの定義。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 以下の要件があります。 ・生物学的原薬及び製剤を製造する人員(保守を含む)は、製造する製品及びその作業に特化した教育訓練及び定期的な再教育訓練を受けること。 ・保守を含む人員に対し、必要に応じて適切なワクチンを接種し、定期的な健康診断受けること。 |
対象医薬品:放射性医薬品 以下の要件があります。 ・放射性製品を製造する保守を含む全従業員は、教育訓練を受けること。 ・主要な装置の保守に係る記録には、日付、時間、担当者の署名、製品名及びロット番号を記載すること。 |
対象医薬品:医療用ガス 以下の要件があります。 ・受託業者を含む保守点検を担当する職員は、教育訓練を受けること。 ・シリンダー、移動型極低温容器及びバルブについて、保守管理及び修理作業を業者に委託する場合には、受託業者との技術協定を含む契約、監査の実施を行うこと。 |
対象医薬品:全般 以下の要件があります。 ・OQが適合して完了することにより、予防保全の要求事項を完成することができる。 ・計画された変更については、メンテナンスに関しても、品質リスク管理を用いること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・無菌性保証プログラムを文書化するとともに、保守管理を文書化を含めること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・滅菌器の不定期及び定期の保守管理活動を記録し、評価すること。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第2章 人員 主要責任者 .2.7 、.2.7(iv)、2.8、2.8 (v)と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第2章 人員 教育訓練 .2.10と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 原則と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第4章 文書化 その他 4.29.、4.31.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第6 章 品質管理 文書化 6.7. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.5と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.11と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:ヒト用生物学的医薬品(原薬及び製剤) 別紙(3) PIC/S GMP ガイドライン アネックス2B ヒト用生物学的医薬品(原薬及び製剤) の製造 パートA 一般的ガイダンス 作業原則 50.と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 人員 17.と同じ内容となります。 文書化 32.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 職員 6.と同じ内容となります。 シリンダーと移動型極低温容器への充填と表示 28.と同じ内容となります。 |
対象医薬品: 全般 別紙(14) PIC/S GMP ガイドライン アネックス15 クオリフィケーション及びバリデーション 3.設備、施設、ユーティリティ及びシステムのクオリフィケーション段階 運転時適格性評価 (OQ) 3.12と同じ内容となります。 11. 変更管理 11.4と同じ内容となります。 |
対象医薬品:対象医薬品:無菌医薬品 別紙(15) PIC/S GMP ガイドライン アネックス17 パラメトリックリリース 4. パラメトリックリリース及び滅菌工程 4.5と同じ内容となります。 |
対象医薬品:対象医薬品:無菌医薬品 別紙(15) PIC/S GMP ガイドライン アネックス17 パラメトリックリリース 4. パラメトリックリリース及び滅菌工程 4.18と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第2章 人員 主要責任者 .2.7 、.2.7(iv)、2.8、2.8 (v)と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第2章 人員。 教育訓練 .2.10と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 原則と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第4章 文書化 その他 4.29.、4.31.と同じ内容となります。 |
対象医薬品: 全般 別紙(1) PIC/S GMP ガイドライン パート1 第6 章 品質管理 文書化 6.7. と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.5と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.11と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 人員 17.と同じ内容となります。 文書化 32.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 職員 6.と同じ内容となります。 シリンダーと移動型極低温容器への充填と表示 28.と同じ内容となります。 |
対象医薬品: 全般 別紙(14) PIC/S GMP ガイドライン アネックス15 クオリフィケーション及びバリデーション 3.設備、施設、ユーティリティ及びシステムのクオリフィケーション段階 運転時適格性評価 (OQ) 3.12と同じ内容となります。 11. 変更管理 11.4と同じ内容となります。 |
対象医薬品:対象医薬品:無菌医薬品 別紙(15) PIC/S GMP ガイドライン アネックス17 パラメトリックリリース 4. パラメトリックリリース及び滅菌工程 4.5と同じ内容となります。 |
対象医薬品:対象医薬品:無菌医薬品 別紙(15) PIC/S GMP ガイドライン アネックス17 パラメトリックリリース 4. パラメトリックリリース及び滅菌工程 4.18と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・製造責任者は部門、建物、設備のメンテナンスについて点検する責任を負うこと。 |
対象医薬品:全般 以下の要件があります。 ・製造業者は、工務、保守、清掃を含む全担当者に対し文書化された手順による訓練をおこなうこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・保守について、SOPおよび記録(適切に完している)が利用できること。 ・重要な機器については、メンテナンス、清掃、修理作業のデータは、作業担当者氏名と日付を含み、適切に保管すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・SOPの定義について。 SOPには機器の操作や保守等についても示すこと。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 2 原則 2.5と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 7 人員 7.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 9 環境及び工程のモニタリング 環境及び工程のモニタリング 9.11と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 用語解説 汚染制御ストラテジー(CCS)と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 日本 | 日本 | 日本 | 米国 | ー | ー | ー | ー | 欧州 | 欧州 | 欧州 | 欧州 | ー | ー | |||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | PIC/S | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | |||||||||||||||||||||||||||||||||||||||||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課長 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | Code of Federal Regulations | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | |||||||||||||||||||||||||||||||||||||||||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 品質リスクマネジメントに関するガイドライン | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(14) PIC/S GMPガイドライン アネックス15 クオリフィケーション及びバリデーション |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 3 MANUFACTURE OF RADIOPHARMACEUTICALS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 6 MANUFACTURE OF MEDICINAL GASES |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 15 QUALIFICATION AND VALIDATION |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 3 Manufacture of Radiopharmaceuticals |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 6 Manufacture of Medicinal Gases |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 15 Qualification and Validation |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
48th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products: main principles WHO Technical Report Series, No. 986, 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||||||||||||||||||||||||||||||||||||||||
| 発行年 | 2011年 | 2006年 | 2025年 | 2012年 | 2013年 | 2015年 | 2023年 | 2023年 | 2023年 | 2023年 | 2023年 | 2023年 | 2008年 | 2010年 | 2015年 | 2014年 | 2022年 | |||||||||||||||||||||||||||||||||||||||||
| 記載内容 | 12. 製造設備及びユーティリティの適格性評価 12.1 一般要件 5) 職員の動き及び無菌製品に係る製品への介入を最小のものとすること。また機器の運転、保全、修理、調整などは、可能な限り重要区域の外から行えるように考慮すること。 |
14. 無菌製造設備の定置清浄化(CIP) 14.5 保守・管理 CIPパラメータである圧力,温度,流量に影響を与えるポンプなどの重要な機器については定期的に保守・管理を行うこと。重要な機器を交換する場合は、同等の性能の機器を選定し交換前と同等なCIPが実施できることを検証し、文書化すること。 |
15. 無菌製造設備の定置蒸気滅菌(SIP) 15. 4 保守・管理 滅菌用蒸気の導入や凝縮水の排水が速やかに行われるようバルブやスチームトラップは定期的に保守を行うこと。SIP対象部の配管の形状やサイズ、スチームの供給条件を変更した場合はバリデーションを実施すること。 |
19. アイソレータシステム/バリアシステム/ブローフィルシール 19.1 アイソレータシステム 適切に設計されたアイソレータは高度な無菌性環境が達成されるが、完全に密閉された空間ではない。従って、薬理活性の高い薬物の製造においては内部を陰圧に保持したアイソレータが用いられることもあるが、通常の無菌医薬品に係る製品の製造においては、内部が陽圧に保持されたアイソレータが用いられる。また、製品の無菌性を高度に保証するためには、HEPAフィルター、グローブ、ハーフスーツ及び各種シール部の保守・点検を含む包括的な予防保全プログラムが必要である |
【参考情報】 A5 ケミカルハザード対策 A5.2 リスクマネジメントプロセス A5.2.2 リスク分析(曝露分析) 患者や製造作業者が,化学物質に曝露されるリスクは,各製造工程のライフサイクル(製造、切り替え洗浄,部品交換,保全などの全製造段階)において取り扱う化学物質の物理的化学的特性(粉体、液体、エアロゾル、粒度、比重、溶解性など)と製造工程自体の特性(開放系/閉鎖系、封じ込め装置、防護具、集塵、定置洗浄、定置湿潤など)に依存する。曝露分析は、定常的におこなわれる製造工程のライフサイクルのみならず、封じ込め装置の破綻といった予測される非定常状態についても実施されなくてはならない。 |
付属書Ⅱ:品質リスクマネジメントの潜在用途 Ⅱ.4 施設、設備、ユーティリティのための品質リスクマネジメント 施設/設備の設計 (略) 関係する設備に対して、予防保全のための適切なメンテナンス方法を決める(例えば必要な予備部品の在庫)。 キャリブレーション/予防保全 適切なキャリブレーション及びメンテナンスの日程を決める。 |
バリア技術 4.20 (略) iアイソレータ b (略)アイソレータについてのCCSのリスク評価を行う際の主要な検討事項には、以下を含めること(ただし、これらに限定されるものではない): (略) 保守管理等の作業のうちアイソレータの最終的なバイオ除染前にそのドアを開けることを要し得るもの。 |
5 設備 5.3 実践可能な限り、設備、備品類及び付帯装置が、作業、保守管理及び修理をクリーンルームの外側で行うことができるように設計され、設置されていること。保守管理をクリーンルーム内で行わなければならず、所要の清浄度及び/又は無菌状態の基準を保つことができないときには、作業区域への立入りを特定の人員に制限する、明確に定められた作業計画書及び保守管理手順を作成する等、予防措置を検討すること。追加的な清浄化、消毒及び環境モニタリングも検討すること。設備の滅菌を要するときには、なるべく組立て完了後に実行すること。 |
5.6 滅菌器、空気処理システム(空気のフィルタ処理を含む) 及び給水システム等の全ての設備が、適格性評価、モニタリング及び計画的な保守管理の対象となっていること。保守管理が完了した際には、その使用再開について承認を受けること。 5.7 製品の無菌性に重要な設備についての計画外の保守管理を行おうとする場合には、製品の無菌性に対する潜在的インパクトの評価を行うとともに記録作成すること。 |
6 ユーティリティ 6.3 ユーティリティは、そのユーティリティのシステムが期待通りに機能することを確保するように設計され、設置され、適格性評価され、稼働し、保守管理され、且つモニターされていること。 |
フォームフィルシール(FFS) 8.101 FFSの重要工程パラメータが、設備の適格性評価の際に決定されていること、また、それには以下を含めるこ と(ただし、これらに限定されるものでない) : i . バリデートされた成形温度(予熱及び冷却の温度を含む)、成形の時間及び圧力(関連する場合)の設定、保守管理及びモニタリング ii. バリデートされた閉塞温度、閉塞物全体に亘る閉塞温度の均一性、閉塞の時間及び圧力( 関連する場合)の設定、保守管理及びモニタリング (略) |
8.104 適切な保守管理手順をリスクに基づいて確立し、それに単位容器の密封の有効性に重要な工作機械の保守管理・検査計画を含めること。潜在的な製品品質の懸念を示唆する問題が特定されたらば、文書化し、原因調査すること。 | 成形同時充填 8.111 粒状樹脂の微粒子・微生物汚染を、当該粒状樹脂の貯蔵、検体採取及び配送のシステムを適切に設計し、管理し且つ保守管理することによって、防止すること。 8.119 適切な保守管理手順をリスクに基づいて確立し、それに単位容器の密封、完全性及び無菌性に重要な項目の保守管理及び検査の計画を含めること。 |
凍結乾燥 8.122 (略) 保守管理又は清掃の後には、再滅菌が行われること。滅菌済みの凍結乾燥機及び付随する設備は、滅菌後の汚染から保護されていること。 |
閉鎖システム 8.128 (略) 無菌操作用の閉鎖システムの設計及び選定は、無菌性の保守管理を確保するものであること。 (略) 8.130 (略) 閉鎖システムが(例:バルク製造ラインの保守管理のため) 開放されるときには、その原材料に適当な等級分け(例: 最終滅菌工程にはグレードC 、無菌操作にはグレードA) がなされた区域内で行う、又は更なる清浄化及び消毒(無菌操作工程の場合においては滅菌) の対象とすること。 |
建物及び設備 全般事項 20. 予防保全、校正、適格性評価プログラムを行い、放射性医薬品の生産に使用される全ての設備及び装置が適切であり適格とされていることを保証しなければならない。これらは、有能な従業員が行い、記録及び日誌を保管しなければならない。 |
建物と設備 設備 15.設備の修理、保守管理(清掃、パージを含む)が、医療用ガスの品質に悪影響を与えてはならない。特に、手順では、システムの完全性が損なわれるような内容を含む修理と保守点検操作の後にとられる対策について記述すること。具体的には、設備の使用可否判定前に最終製品の品質に悪影響を与えるような汚染がないことを示すこと。記録は保存すること。 |
製造 シリンダーと移動型極低温容器への充填と表示 25 シリンダー、移動型極低温容器及びバルブは、初回製造での使用開始前に確認し、適切に保守管理すること。医療機器が適合性評価手順に従って処理された場合は、保守管理は医療機器製造業者の指示書の内容を遵守すること。 26.確認及び保守管理操作は、医薬品の品質及び安全性に影響を与えてはならない。容器の耐圧試験に使用される水は、少なくとも飲料水の品質であること。 |
3.設備、施設、ユーティリティ及びシステムのクオリフィケーション段階 設備据付時適格性評価(IQ) 3.9 IQは、これらに限定されないが以下を含まなければならない: iii. 供給業者の操作及び作業説明書、及びメンテナンス要求事項の収集と確認 |
Subpart D--Equipment Sec. 211.67 Equipment cleaning and maintenance. (a) Equipment and utensils shall be cleaned, maintained, and, as appropriate for the nature of the drug, sanitized and/or sterilized at appropriate intervals to prevent malfunctions or contamination ・・・ (b) Written procedures shall be established and followed for cleaning and maintenance of equipment, (略) (1) Assignment of responsibility for cleaning and maintaining equipment; (2) Maintenance and cleaning schedules, including, where appropriate, sanitizing schedules; (3) A description in sufficient detail of the methods, equipment, and materials used in cleaning and maintenance operations, ・・・(略) |
(c) Records shall be kept of maintenance, cleaning, sanitizing, and inspection as specified in 211.180 and 211.182. | BARRIER TECHNOLOGIES 4.20 (略) i. Isolators: ibKey considerations when performing the risk assessment for the CCS of an isolator should include (but are not limited to); (略) maintenance that may require the doors to be opened prior to the final bio-decontamination of the isolator. |
EQUIPMENT 5.3 As far as practicable, equipment, fittings and services should be designed and installed so that operations, maintenance, and repairs can be performed outside the cleanroom. If maintenance has to be performed in the cleanroom, and the required standards of cleanliness and/or asepsis cannot be maintained, then precautions such as restricting access to the work area to specified personnel, generation of clearly defined work protocols and maintenance procedures should be considered. Additional cleaning, disinfection and environmental monitoring should also be considered. If sterilisation of equipment is required, it should be carried out, wherever possible, after complete reassembly. |
5.6 All equipment such as sterilisers, air handling systems (including air filtration) and water systems should be subject to qualification, monitoring and planned maintenance. Upon completion of maintenance, their return to use should be approved. 5.7 Where unplanned maintenance of equipment critical to the sterility of the product is to be carried out, an assessment of the potential impact to the sterility of the product should be performed and recorded. |
6 Utilities 6.3 Utilities should be designed, installed, qualified, operated, maintained and monitored in a manner to ensure that the utility system functions as expected. |
FORM-FILL-SEAL (FFS) 8.101 Critical process parameters for FFS should be determined during equipment qualification and should include, but are not limited to: ii. setting, maintenance and monitoring of validated forming temperatures (including pre-heating and cooling), forming times and pressures as relevant; iii. setting, maintenance and monitoring of validated sealing temperatures, sealing temperature uniformity across the seal, sealing times and pressures as relevant; |
8.104 Appropriate maintenance procedures should be established based on risk, and include maintenance and inspection plans for tooling critical to the effectiveness of unit sealing. Any issues identified that indicate a potential product quality concern should be documented and investigated. | BLOW-FILL-SEAL 8.111 Particulate and microbial contamination of the polymer granulate should be prevented by appropriate design, control, and maintenance of the polymer granulate storage, sampling and distribution systems. 8.119 Appropriate maintenance procedures should be established based on risk, and include maintenance and inspection plans for items critical to unit sealing, integrity and sterility. |
LYOPHILIZATION 8.122 (略) Re-sterilisation should be performed following maintenance or cleaning. Sterilised lyophilizers and associated equipment should be protected from contamination after sterilisation. |
CLOSED SYSTEMS 8.128 (略) The design and selection of any closed system used for aseptic processing should ensure maintenance of sterility.(略) 8.130 (略) If the closed system is opened (e.g. for maintenance of a bulk manufacturing line) then this should be performed in a classified area appropriate to the materials (e.g. grade C for terminal sterilisation processes, or grade A for aseptic processing) or be subject to further cleaning and disinfection (and sterilisation in case of aseptic processes). |
PREMISES AND EQUIPMENT General 20. Preventive maintenance, calibration and qualification programmes should be operated to ensure that all facilities and equipment used in the manufacture of radiopharmaceutical are suitable and qualified. These activities should be carried out by competent personnel and records and logs should be maintained. |
PREMISES AND EQUIPMENT Equipment 15.Repair and maintenance operations (including cleaning and purging) of equipment, should not adversely affect the quality of the medicinal gases. In particular, procedures should describe the measures to be taken after repair and maintenance operations involving breaches of the system’s integrity. Specifically it should be demonstrated that the equipment is free from any contamination that may adversely affect the quality of the finished product before releasing it for use. Records should be maintained. |
PRODUCTION Filling and labelling of cylinders and mobile cryogenic vessels 25. Cylinders, mobile cryogenic vessels and valves should be checked before first use in production, and should be properly maintained. Where medical devices have gone through a conformity assessment procedure1, the maintenance should address the medical device manufacturer’s instructions. 26. Checks and maintenance operations should not affect the quality and the safety of the medicinal product. The water used for the hydrostatic pressure testing carried out on cylinders should be at least of drinking quality. |
3.QUALIFICATION STAGES FOR EQUIPMENT, FACILITIES, UTILITIES AND SYSTEMS. Installation qualification (IQ) 3.9 IQ should include, but is not limited to the following: iii. Collection and collation of supplier operating and working instructions and maintenance requirements; |
Barrier Technolog 4.20 (略) i. Isolators: b. Key considerations when performing the risk assessment for the CCS of an isolator should include (but are not limited to); (略) maintenance that may require the doors to be opened prior to the final bio-decontamination of the isolator. |
5 Equipment 5.3 As far as practicable, equipment, fittings and services should be designed and installed so that operations, maintenance, and repairs can be performed outside the cleanroom. If maintenance has to be performed in the cleanroom, and the required standards of cleanliness and/or asepsis cannot be maintained, then precautions such as restricting access to the work area to specified personnel, generation of clearly defined work protocols and maintenance procedures should be considered. Additional cleaning, disinfection and environmental monitoring should also be considered. If sterilisation of equipment is required, it should be carried out, wherever possible, after complete reassembly. |
5.6 All equipment such as sterilisers, air handling systems (including air filtration) and water systems should be subject to qualification, monitoring and planned maintenance. Upon completion of maintenance, their return to use should be approved. 5.7 Where unplanned maintenance of equipment critical to the sterility of the product is to be carried out, an assessment of the potential impact to the sterility of the product should be performed and recorded. |
6 Utilities 6.3 Utilities should be designed, installed, qualified, operated, maintained and monitored in a manner to ensure that the utility system functions as expected. |
Form-Fill-Seal (FFS) 8.101 Critical process parameters for FFS should be determined during equipment qualification and should include, but are not limited to: ii. Setting, maintenance and monitoring of validated forming temperatures (including pre-heating and cooling), forming times and pressures as relevant. iii. Setting, maintenance and monitoring of validated sealing temperatures, sealing temperature uniformity across the seal, sealing times and pressures as relevant. |
8.104 Appropriate maintenance procedures should be established based on risk, and include maintenance and inspection plans for tooling critical to the effectiveness of unit sealing. Any issues identified that indicate a potential product quality concern should be documented and investigated. | Blow-Fill-Seal 8.111 Particulate and microbial contamination of the polymer granulate should be prevented by appropriate design, control, and maintenance of the polymer granulate storage, sampling and distribution systems. 8.119 Appropriate maintenance procedures should be established based on risk, and include maintenance and inspection plans for items critical to unit sealing, integrity and sterility. |
Lyophilization 8.122 (略) Re-sterilisation should be performed following maintenance or cleaning. Sterilised lyophilizers and associated equipment should be protected from contamination after sterilisation. |
Closed systems 8.128 (略) The design and selection of any closed system used for aseptic processing should ensure maintenance of sterility (略) 8.130 (略) If the closed system is opened (e.g. for maintenance of a bulk manufacturing line) then this should be performed in a classified area appropriate to the materials (e.g. grade C for terminal sterilisation processes, or grade A for aseptic processing) or be subject to further cleaning and disinfection (and sterilisation in case of aseptic processes). |
Premises and equipment 20. Preventive maintenance, calibration and qualification programmes should be operated to ensure that all facilities and equipment used in the manufacture of radiopharmaceutical are suitable and qualified.These activities should be carried out by competent personnel and records and logs should be maintained. |
Equipment 15. Repair and maintenance operations (including cleaning and purging) of equipment, should not adversely affect the quality of medicinal gases. In particular, procedures should describe the measures to be taken after repair and maintenance operations involving breaches of the system’s integrity. Specifically it should be demonstrated that the equipment is free from any contamination that may adversely affect the quality of the finished product before releasing it for use. Records should be maintained. |
Production Filling and labelling of cylinders and mobile cryogenic vessels 25. Cylinders, mobile cryogenic vessels and valves should be checked before first use in production, and should be properly maintained. Where CE marked medical devices are used, the maintenance should address the medical device manufacturer’s instructions. 26. Checks and maintenance operations should not affect the quality and the safety of the medicinal product. The water used for the hydrostatic pressure testing carried out on cylinders should be at least of drinking quality |
3. QUALIFICATION STAGES FOR EQUIPMENT, FACILITIES, UTILITIES AND SYSTEMS. Installation qualification (IQ) 3.9. IQ should include, but is not limited to the following: iii. Collection and collation of supplier operating and working instructions and maintenance requirements; |
13. Equipment 13.1 Equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. The layout and design of equipment must aim to minimize the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt, and, in general, any adverse effect on the quality of products. |
16. Good practices in production 16.24 Repair and maintenance operations should not present any hazard to the quality of the products. |
Barrier technologies 4.20 (略) i. Isolators: Key considerations when performing the risk assessment for the CCS of an isolator should include (略) maintenance that may require the doors to be opened prior to the final biodecontamination of the isolator. |
5. Equipment 5.3 As far as practicable, equipment, fittings and services should be designed and installed so that operations, maintenance, and repairs can be performed outside the cleanroom. If maintenance has to be performed in the cleanroom, and the required standards of cleanliness or asepsis cannot be maintained, then precautions such as restricting access to the work area to specified personnel and generation of clearly defined work protocols and maintenance procedures should be considered. Additional cleaning, disinfection and environmental monitoring should also be performed where appropriate. If sterilization of equipment is required, it should be carried out, wherever possible, after complete reassembly. |
5.6 All equipment, such as sterilizers, air handling systems (including air filtration systems) and water systems, should be subject to qualification, monitoring and planned maintenance. Upon completion of maintenance or repairs, their return to use should be approved. 5.7 Where unplanned maintenance of equipment critical to the sterility of the product is to be carried out, an assessment of the potential impact to the sterility of the product should be performed and recorded. |
6. Utilities 6.3 Utilities should be designed, installed, qualified, operated, maintained and monitored in a manner that ensures that the utility system functions as expected. |
Form-fill-seal (FFS) 8.101 Critical process parameters for FFS should be determined during equipment qualification and should include: ii. setting, maintenance and monitoring of validated forming temperatures (including preheating and cooling), forming times and pressures, as relevant; iii. setting, maintenance and monitoring of validated sealing temperatures, sealing temperature uniformity across the seal, sealing times and pressures, as relevant; (略) |
8.104 The appropriate maintenance procedures should be established based on risk, and should include maintenance and inspection plans for tooling critical to the effectiveness of unit sealing. Any issues identified that indicate a potential product quality concern should be documented and investigated. | Blow-fill-seal (BFS) 8.111 Particulate and microbial contamination of the polymer granulate should be prevented by the appropriate design, control and maintenance of the polymer granulate storage, sampling and distribution systems. 8.119 The appropriate maintenance procedures should be established based on risk, and should include maintenance and inspection plans for items critical to unit sealing, integrity and sterility. |
Lyophilization 8.122 (略) Resterilization should be performed following maintenance or cleaning. Sterilized lyophilizers and associated equipment should be protected from contamination after sterilization. |
Closed systems 8.128 (略) The design and selection of any closed system used for aseptic processing should ensure that sterility is achieved and maintained. (略) If the closed system is opened (for example, for maintenance of a bulk manufacturing line), then this should be performed in a classified area appropriate to the materials (for example, grade C for terminal sterilization processes or grade A for aseptic processing) or be subject to further cleaning and disinfection (and sterilization in the case of aseptic processes). |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・無菌製品への介入は最小とすること。 ・機器の運転、保全は、可能な限り重要区域の外から行えること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・CIPパラメータで重要な機器は定期的に保守・管理を行うこと。 ・重要な機器を交換する場合は交換前と同等なCIPが実施できること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・滅菌用蒸気や凝縮水の排水が速やかに行われるよう定期的に保守を行うこと。 ・SIP対象部の供給条件を変更した場合、バリデーションを実施すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・製品の無菌性を高度に保証するためには、アイソレータの各種シール部の保守・点検を含む包括的な予防保全プログラムが必要である。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・定常的におこなわれる製造工程のライフサイクル(部品交換,保全などの全製造段階を含む。)にて、リスク分析(曝露分析)を実施すること。 |
対象医薬品:全般 以下の要件があります。 ・関係する設備に対して、予防保全のための適切なメンテナンス方法(例:必要な予備部品の在庫)を決めること。 ・予防保全について: 適切なメンテナンスの日程を決めること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・アイソレータの保守管理で最終的なバイオ除染前にそのドアを開ける場合は、CCSのリスク評価に含めること。 *用語についての補足:「CCS」は 空調設備 002:差圧 のアネックス 1の用語解説を参照ください。 *用語についての補足:「アイソレータ」は 空調設備 003:清浄度 のアネックス 1の用語解説を参照ください。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・保守管理及び修理は、可能な限り清浄区域外からできるよう設計し、設置すること。 ・清浄区域内で保守管理を行う場合、予防措置、追加的な清浄化、消毒及び環境モニタリングを検討すること。滅菌が必要な場合は、完全に組み立てが終了してから行うこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・全ての設備は計画的維持管理の対象とすること。 ・無菌性に重要な設備の計画外の保守管理を行う場合は、製品の無菌性に対する潜在的インパクトの評価を行い、記録すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・ユーティリティについて、保守管理すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・フォームフィルシール(FFS)について、 適格性評価の際に決定されている重要工程パラメータに、保守管理を含めること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・フォームフィルシール(FFS)について、 単位容器の密封の有効性に重要な工作機械を含む適切な保守管理手順をリスクに基づいて確立すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・成形同時充填について、保守管理することで粒状樹脂の微粒子・微生物汚染を防止すること。 ・成形同時充填について、単位容器の密封、完全性及び無菌性に重要な項目を含む適切な保守管理手順をリスクに基づいて確立すること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・凍結乾燥機について、保守管理後に再滅菌が行われること。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・閉鎖システムについて、無菌性の保守管理を確保するものであること。 ・閉鎖システムについて、保守管理のために開放する場合は、原材料に適当な等級域内で行うこと。更なる清浄化及び消毒の対象とすること。 |
対象医薬品:放射性医薬品 以下の要件があります。 ・全ての設備及び装置について、予防保全を行うこと。 |
対象医薬品:医療用ガス 以下の要件があります。 ・修理や保守作業が、医療用ガスの品質に影響を与えないこと。 ・手順書にシステムの完全性が損なわれるような内容を含む修理と保守点検操作の後にとられる対策について記述すること。 |
対象医薬品:医療用ガス 以下の要件があります。 ・極低温の液化ガスの移送と配送のシリンダー、移動型極低温容器及びバルブについて、初回製造での使用開始前に確認し、適切に保守管理すること。 ・シリンダーと移動型極低温容器について、保守作業が、医療用ガスの品質に影響を与えないこと。 |
対象医薬品:全般 以下の要件があります。 ・設備据付時適格性評価(IQ)に、供給業者の操作及び作業説明書、及びメンテナンス要求事項の収集と確認を含めること。 |
対象医薬品:全般 以下の要件があります。 ・適切な間隔での設備等の保守を行うこと。 製造設備の衛生管理手順書について。 ・使用する器具を含む設備の保守を行うための手順書を作成し、これを遵守すること。 ・設備保全の責任者の割り当てること。 ・保守日程計画を作成すること。(該当する場合には、清浄化作業日程計画を含むこと。) ・保守作業で使用する方法、設備、及び物品の詳細な記述を含むこと。 ・保守を保証するため必要な機器の分解と組み付け方法の詳細な記述を含むこと。 |
対象医薬品:全般 以下の要件があります。 ・§211.180および§211.182に規定されているように、保守の記録を保持していること。 注釈:Sec.211.180 General requirements.(一般要件) Sec.211.182 Equipment cleaning and use log.(装置の清浄化および使用ログ) |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン 4 建物 アネックス1 無菌医薬品の製造 バリア技術 4.20 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.6、5.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 6.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 フォームフィルシール(FFS) 8.101と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 フォームフィルシール(FFS) 8.104と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 成形同時充填 8.111、8.119と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 凍結乾燥 8.122と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 閉鎖システム 8.128、8.130と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 建物及び設備 全般事項 20.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 建物と設備 設備 15.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 製造 シリンダーと移動型極低温容器への充填と表示 25.、26.と同じ内容となります。 |
対象医薬品: 全般 別紙(14) PIC/S GMP ガイドライン アネックス15 クオリフィケーション及びバリデーション 3.設備、施設、ユーティリティ及びシステムのクオリフィケーション段階 設備据付時適格性評価(IQ) 3.9 と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン 4 建物 アネックス1 無菌医薬品の製造 バリア技術 4.20 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.6、5.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 6.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 フォームフィルシール(FFS) 8.101と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 フォームフィルシール(FFS) 8.104と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 成形同時充填 8.111、8.119と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 凍結乾燥 8.122と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 閉鎖システム 8.128、8.130と同じ内容となります。 |
対象医薬品:放射線医薬品 別紙(4) PIC/S GMP ガイドライン アネックス3 放射性医薬品の製造 建物及び設備 全般事項 20.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 建物と設備 設備 15.と同じ内容となります。 |
対象医薬品:医療用ガス 別紙(5) PIC/S GMP ガイドライン アネックス6 医療用ガスの製造 製造 シリンダーと移動型極低温容器への充填と表示 25.、26.と同じ内容となります。 |
対象医薬品: 全般 別紙(14) PIC/S GMP ガイドライン アネックス15 クオリフィケーション及びバリデーション 3.設備、施設、ユーティリティ及びシステムのクオリフィケーション段階 設備据付時適格性評価(IQ) 3.9 と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・設備は製剤操作の実施に適した保守をすること。 ・設備の配置と設計は、ミスするリスクの最小化とコンタミと汚れの蓄積をさけるような清掃と保全を実施することで製品品質に悪影響を与えないことを目的とすること。 |
対象医薬品:全般 以下の要件があります。 ・修理と保守作業が製品品質を危険にさらすようなことがあってはならない。 |
別紙(2) PIC/S GMP ガイドライン 4 建物 アネックス1 無菌医薬品の製造 バリア技術 4.20 i bと同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 5 設備 5.6、5.7と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 6.3と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 フォームフィルシール(FFS) 8.101と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 フォームフィルシール(FFS) 8.104と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 成形同時充填 8.111、8.119と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 凍結乾燥 8.122と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 閉鎖システム 8.128、8.130と同じ内容となります。 |
| 国 | 日本 | ー | 欧州 | ー | ー | |||
|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION | WHO | WHO | |||
| 分 類 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | |||
| 法規類 タイトル |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 52nd report Annex 8 Guidelines on heating, ventilation and air-conditioning systems for non-sterile pharmaceutical products WHO Technical Report Series, No. 1010 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||
| 発行年 | 2025年 | 2023年 | 2023年 | 2018年 | 2022年 | |||
| 記載内容 | クリーンルーム及び清浄空気設備の適格性評価 4.32 クリーンルーム及び清浄空気設備の適格性再評価が、所定の手順に従って定期的に行われていること。その適格性再評価には最低限、以下を含めること: (略) 変更管理プロセスを通じて、変更の重大性を判定すること。検討すべき変更の事例には以下が含まれるが、これらに限定されるものではない: (略) iii設備の稼働に影響を及ぼす特別な保守管理( 例: 最終フィルタの交換) |
CLEANROOM AND CLEAN AIR EQUIPMENT QUALIFICATION 4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include at a minimum the following: (略) Examples of changes to be considered include but are not limited to the following: (略) iii. special maintenance which affects the operation of the installation (e.g. change of final filters). |
Cleanroom and clean air equipment qualification 4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include at a minimum the following: (略) Examples of changes to be considered include but are not limited to the following: (略) iii. Special maintenance which affects the operation of the installation (e.g. change of final filters). |
13. Maintenance 13.1 Operation and maintenance (O&M) manuals, procedures and records should be available and kept up to date with details of any system revisions made. 13.2 O&M manuals, schematic drawings, protocols and reports should be maintained as reference documents for any future changes and upgrades to the system. |
13.3 The O&M manuals may typically contain the following information:
・system description; ・operating instructions; ・troubleshooting; ・commissioning data; ・maintenance instructions; ・list of equipment suppliers; ・spare parts list; ・equipment data/capacity schedules; ・supplier’s literature; ・control system description; ・electrical drawings; ・as-built drawings. |
13.4 There should be a planned preventive maintenance programme for the HVAC system. The details of this programme should be commensurate with the criticality of the system and components. 13.5 Maintenance activities should not have any negative impact on product quality and should normally be scheduled to take place at appropriate times, for example, outside production hours. In case of system stoppages, appropriate quality management system procedures should be followed. Where necessary, the root cause and impact should be assessed and appropriate corrective and preventive action taken. Where necessary, qualification or requalification should be considered. |
13.6 HEPA filters should be changed by a competent person and this should be followed by installed filter leakage testing. 13.7 Records should be kept for a sufficient period of time. |
Cleanroom and clean air equipment qualification 4.32 The requalification of cleanrooms and clean air equipment should be carried out periodically following defined procedures. The requalification should include, at a minimum, the following: (略) Examples of changes requiring requalification include the following: (略) iii. special maintenance that affects the operation of the installation (such as a change of final filters). |
| 概 要 | 対象医薬品:無菌医薬品 以下の要件があります。 ・クリーンルーム及び清浄空気設備の適格性再評価に、 最終フィルタの交換等の設備の稼働に影響を及ぼす特別な保守管理を含めること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32と同じ内容となります。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調システムの機械操作と保守の手順書と記録書は整備、保管し、必要に応じて改訂すること。 ・空調システムの機械操作と保守のマニュアルと概略図、計画書、報告書は必要に応じて改訂すること。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調システムの機械操作と保守の手順書には下記のような情報を記載すること。 システムの状態、操作手順、異常時の対策、試運転時のデータ、保全手順、設備メーカーのリスト、予備部品リスト、設備データ、能力表、機械メーカー文書、制御系記述、電気図面、完成図。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・空調システムの設備に関する計画的な予防保全を実施すること。 ・保守活動は製品品質に悪影響を与えないよう、製造時間外に設定すること。設備が停止した場合には、原因究明や影響評価が求められ、是正や予防の措置が取られることがあり、クオリフィケーションや再クオリフィケーションが必要になることがある。 |
対象医薬品:非無菌医薬品 以下の要件があります。 ・HEPAフィルターは適格な担当者が交換し、更新後はリークテストを実施すること。 ・記録文書は十分な期間、保管すること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 クリーンルーム及び清浄空気設備の適格性評価 4.32と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | ー | ー | 欧州 | 欧州 | ー | ー | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | |||
| 分 類 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | GMP | GMP | GMP | GMP | GMP | GMP | |||
| 法規類 タイトル |
無菌操作法による無菌医薬品の製造に関する指針 | 「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(8) PIC/S GMP ガイドライン アネックス9 液剤、クリーム剤及び軟膏剤の製造 |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 9 MANUFACTURE OF LIQUIDS, CREAMS AND OINTMENTS |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 9 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations 55th report Annex 3 Good manufacturing practices: water for pharmaceutical use WHO Technical Report Series, No. 1033, 2021 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
|||
| 発行年 | 2011年 | 2025年 | 2012年 | 2023年 | 2023年 | 2023年 | 2011年 | 2021年 | 2022年 | |||
| 記載内容 | 【参考情報】 A2 製薬用水 A2.2 製薬用水のバリデーション 8) 保全プログラム 既にバリデーションが実施された製薬用水設備について、工程管理の定期照査について手順書等を適切に作成し実施するとともに、定期的再バリデーション(再校正を含む。)についてバリデーション実施計画書(スケジュールを含む。)及び手順書等を適切に作成の上実施し、その結果を踏まえ必要に応じ予防措置又は是正措置(適切な変更管理を含む。)を採ること。. |
A2.5 製薬用設備の維持管理 水の設備を管理状態に保つことを確実とするために予防的保全計画を確立すべきであり、そのプログラムには,次のような事項を含めること。 1) 設備を運転するための方法 2) 重要な品質特性及び運転条件のモニタリングプログラム 3) 定期的なサニタイゼーションスケジュール 4) 設備構成要素の予防的維持管理及び校正 5) 設備及びその運転条件に対する変更管理 6) 装置の一時停止時及び再稼動時においての手順 特に超ろ過膜処理装置に関する手順については,膜面の劣化によるリークの恐れという観点から十分な配慮を行うこと 7) 用水設備及びメンテナンスの理解 |
A2.6 変更管理 設備について改造あるいは増設を行った場合又はその運転方法を変更した場合は、その変更が製薬用水の品質に及ぼす影響を評価すること.その変更により、設備の再評価が必要と判断される場合は、その内容に応じ適切なバリデーションを行う必要がある。保全プログラムの一環として、これら変更時の管理方法を定めておくこと。これらの変更管理手順は、バリデーションと保全の一環として,プログラムに定めておくこと。 |
給水システム 6.7 微生物汚染を防ぎ、適切な品質の水の信頼できる供給源を確保するように、水処理設備及び配水システムが設計され、組み立てられ、据え付けられ、就役され、適格性評価され、モニターされ、且つ保守管理されていること。微粒子、微生物の汚染/増殖及びエンドトキシン/発熱性物質の存在のリスクを最小化するよう措置を講じること(例: 配管に傾斜を付けて完全に排水できるようにして、デッドレグをなくす) 。システム中にフィルタが含まれている場合には、そのモニタリング及び保守管理に特別な注意を払うこと。生産された水は、関連する薬局方の現行のモノグラフに適合すること。 |
製造 4. 製造において使用する水の化学的及び微生物学的品質を特定し、モニターする必要がある。微生物増殖のリスクを避けるため、水システムの保守点検に注意を払う必要がある。水システムの化学的消毒後にはバリデーションを実施済のフラッシング手順に従って消毒薬が効果的に除去されたことを保証する必要がある。 |
WATER SYSTEMS 6.7 Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination/proliferation and endotoxin/pyrogen (e.g. sloping of piping to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant Pharmacopeia. |
PRODUCTION 4. The chemical and microbiological quality of water used in production should be specified and monitored. Care should be taken in the maintenance of water systems in order to avoid the risk of microbial proliferation. After any chemical sanitization of the water systems, a validated flushing procedure should be followed to ensure that the sanitising agent has been effectively removed. |
Water systems 6.7 Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination/proliferation and endotoxin/pyrogen (e.g. sloping of piping to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant Pharmacopeia. |
Production 4. The chemical and microbiological quality of water used in production should be specified and monitored. Care should be taken in the maintenance of water systems in order to avoid the risk of microbial proliferation. After any chemical sanitisation of the water systems, a validated flushing procedure should be followed to ensure that the sanitising agent has been effectively removed. |
2. Background to water requirements and uses 2.3 To reduce the risks associated with the production, storage and distribution of water, and considering the properties and use, it is essential: ■to ensure the appropriate design, installation, operation and maintenance of WPU, pre-treatment, treatment, storage and distribution systems; 13. Maintenance of water systems 13.2 The maintenance programme should take into account at least the following: (略); ■ preventive maintenance and maintenance plan and instructions, including cleaning after maintenance; ■a record and review of problems and faults during maintenance |
14. System reviews 14.3 Examples of matters to be included in the review are :(略); ■ maintenance and calibration history; |
Water systems 6.7 (略) Water treatment plant and distribution systems should be designed, constructed, installed, commissioned, qualified, monitored and maintained to prevent microbiological contamination and to ensure a reliable source of water of an appropriate quality. Measures should be taken to minimize the risk of presence of particulates, microbial contamination and proliferation, and endotoxin/pyrogen (for example, by sloping pipes to provide complete drainage and the avoidance of dead legs). Where filters are included in the system, special attention should be given to their monitoring and maintenance. Water produced should comply with the current monograph of the relevant pharmacopoeia. |
| 概 要 | 対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・保全プログラムとして、工程管理の定期照査および定期的再バリデーションについて手順書等の適切な作成を実施すること。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・予防的保全計画を確立すること。 ・計画に含めるべき事項が示されていること。 ・超ろ過膜処理装置に関する手順については,膜面の劣化によるリークの恐れという観点から十分な配慮を行うこと。 |
対象医薬品:無菌操作法による無菌医薬品 以下の要件があります。 ・変更時の管理手順、方法を、保全プログラムの一環として定めておくこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・水処理設備及び配水システムについて、保守管理すること。 |
対象医薬品:液剤、クリーム剤及び軟膏剤 以下の要件があります。 ・水システムの微生物増殖のリスクを避けるため、保守点検時に注意すること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.3と同じ内容となります。 |
対象医薬品:液剤、クリーム剤及び軟膏剤 別紙(8) PIC/S GMP ガイドライン アネックス9 液剤、クリーム剤及び軟膏剤の製造 製造 4..と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.3と同じ内容となります。 |
対象医薬品:液剤、クリーム剤及び軟膏剤 別紙(8) PIC/S GMP ガイドライン アネックス9 液剤、クリーム剤及び軟膏剤の製造 製造 4..と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・製造用水システムの製造、貯留、配水について、リスクを低減し、以下の特性を考慮すること。 ー保守 ・製造用水システムの保全プログラムには、下記の事項を考慮すること。 ー予防保全、保守後のクリーニングを含む保全計画と手順 保守中の問題と欠陥の記録と照査 |
対象医薬品:全般 以下の要件があります。 ・製造用水システムのシステム照査には以下を含むこと。 ー保守と校正履歴 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 6 ユーティリティ 給水システム 6.3と同じ内容となります。 |
| 国 | 日本 | ー | 欧州 |
|---|---|---|---|
| 発行機関 | 厚生労働省 | PIC/S | EUROPEAN COMMISSION |
| 分 類 | 医薬食品局完成指導・麻薬対策課 | GMP | GMP |
| 法規類 タイトル |
「PIC/SのGMPガイドラインを活用する際の考え方について」 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 11 MANUFACTURE OF STERILE COMPUTERISED SYSTEMS |
EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use ANNEX 11 Computerised Systems |
| 発行年 | 2012年 | 2023年 | 2011年 |
| 記載内容 | 用語 ライフサイクル: 設計、規格、プログラム作成、試験、設置、操作、保守管理を含めた、初期の要求事項から廃棄までのシステムの耐用期間における全段階。 システムオーナー: コンピュータ化システム及びシステム上に存在するデータのセキュリティの有用性、及び保守管理に対して責任を負う人物。 |
GLOSSARY Life cycle All phases in the life of the system from initial requirements until retirement including design, specification, programming, testing, installation, operation, and maintenance. System owner The person responsible for the availability, and maintenance of a computerised system and for the security of the data residing on that system. |
Glossary Life cycle: All phases in the life of the system from initial requirements until retirent including design, specification, programming, testing, installation, operation, and maintenance. System owner: The person reponsible for the availability, and maintenance of a computersed system and for the security of the data residing on that system. |
| 概 要 | 対象医薬品:全般 以下の要件があります。 ・用語の定義の中で、ライフサイクルには、保守管理が含まれる。 また、システムオーナーは、保守管理に対して責任を負う人物である。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 用語と同じ内容となります。 |
対象医薬品:全般 別紙(10) PIC/S GMP ガイドライン アネックス11 コンピュータ化システム 用語と同じ内容となります。 |
| 国 | 日本 | 日本 | 日本 | 米国 | ー | ー | ー | 欧州 | 欧州 | 欧州 | ー | ー | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 発行機関 | 厚生労働省 | 厚生労働省 | 厚生労働省 | U.S. Food and Drug Administration | PIC/S | PIC/S | PIC/S | EUROPEAN COMMISSION | EUROPEAN COMMISSION | EUROPEAN COMMISSION | WHO | WHO | ||||||||||||||
| 分 類 | 医薬・生活衛生局監視指導・麻薬対策課事務連絡 | 医薬局監視指導・麻薬対策課事務連絡 | 医薬食品局監視指導・麻薬対策課事務連絡 | Code of Federal Regulations | GMP | GMP | GMP | GMP | GMP | GMP | GMP | GMP | ||||||||||||||
| 法規類 タイトル |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(1)PIC/S GMP ガイドライン パート 1 |
「PIC/S のGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(2)PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 |
「PIC/SのGMPガイドラインを活用する際の考え方について」の一部改正について 別紙(6) PIC/S GMP ガイドライン アネックス7 |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES SUBCHAPTER C--DRUGS: GENERAL PART 211 CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS PART I | GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS |
GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS ANNEX 7 MANUFACTURE OF HERBAL MEDICINAL PRODUCTS |
EU Guidelines to Good Manufacturing Practice
Medicinal Products for Human and Veterinary Use Part 1 |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 1 Manufacture of Sterile Medicinal Products |
EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use ANNEX 7 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
48th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products: main principles WHO Technical Report Series, No. 986, 2014 |
WHO Expert Committee on Specifications for Pharmaceutical Preparations
56th report Annex 2 WHO good manufacturing practices for sterile pharmaceutical products WHO Technical Report Series, No. 1044, 2022 |
||||||||||||||
| 発行年 | 2017年 | 2025年 | 2013年 | 2018年 | 2021年 | 2023年 | 2023年 | 2015年 | 2023年 | 2009年 | 2014年 | 2022年 | ||||||||||||||
| 記載内容 | 第3 章建物及び設備 原則 実施される作業にふさわしいように、建物及び装置を配置し、設計し、建造し、供用し、保守管理しなければならない。その配置及び設計は、過誤のリスクを最小にすることを目途とするとともに、交叉汚染、じん埃又は汚れの蓄積及び( 一般的に)製品品質への悪影響を回避するために、有効な洗浄及び保守管理を可能とするものでなければならない。 |
第3章 建物及び設備 建物 全般事項 3.2. 補修及び保守管理作業が製品の品質にいかなる危険も示さないことを保証するよう、建物は注意深く維持管理されること。詳細な文書化された手順に従い清掃し、該当する場合には消毒しなければならない。 |
第3章 建物及び設備 建物 製造区域 3.10. 配管、照明取り付け具、換気及び他のサービス供給箇所は、清掃しにくい窪みの形成を回避するように設計及び配置すること。保守の目的の為、できる限り製造区域外から到達可能でなければならない。 |
第3章 建物及び設備 建物 付随区域 3.32. 保守管理の作業場は、製造区域からできるだけ離されていること。部品及び工具が製造区域で保管される場合には常に、それらはその用途に専用の部屋又はロッカーの中で保管されなければならない。 |
第3章 建物及び設備 設備 3.35. 修復及び保守管理作業は製品品質に対し、いかなる危険も示さないこと。 |
4 建物 4.9 (中略)低い等級のクリーンルーム内の床排水口には、逆流を防止する設計のトラップ又はウォータシールが取り付けられ、定期的に清浄化され、消毒され、保守管理されていること。 |
植物性医薬品の製造 建物 保管区域 3. 保管区域の清浄性及び的確な保守管理には特別な注意を払うこと。特に埃が発生する場合は、注意すること。 |
Subpart C--Buildings and Facilities Sec. 211.58 Maintenance. Any building used in the manufacture, processing, packing, or holding of a drug product shall be maintained in a good state of repair. | CHAPTER 3 PREMISES AND EQUIPMENT PRINCIPLE Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt and, in general, any adverse effect on the quality of products. |
CHAPTER 3 PREMISES AND EQUIPMENT PREMISES General 3.2. Premises should be carefully maintained, ensuring that repair and maintenance operations do not present any hazard to the quality of products. They should be cleaned and, where applicable, disinfected according to detailed written procedures. |
CHAPTER 3 PREMISES AND EQUIPMENT Production Areas 3.10 Pipework, light fittings, ventilation points and other services should be designed and sited to avoid the creation of recesses which are difficult to clean. As far as possible, for maintenance purposes, they should be accessible from outside the manufacturing areas. |
CHAPTER 3 PREMISES AND EQUIPMENT Ancillary Areas 3.32. Maintenance workshops should as far as possible be separated from production areas. Whenever parts and tools are stored in the production area, they should be kept in rooms or lockers reserved for that use. |
CHAPTER 3 PREMISES AND EQUIPMENT EQUIPMENT 3.35. Repair and maintenance operations should not present any hazard to the quality of the products. |
4 Premises 4.9 (略) Floor drains in lower grade cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be regularly cleaned, disinfected and maintained. |
PREMISES Storage areas 3. Special attention should be paid to the cleanliness and good maintenance of the storage areas particularly when dust is generated. |
Chapter 3: Premises and Equipment PRINCIPLE Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out. Their layout and design must aim to minimise the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt and, in general, any adverse effect on the quality of products. |
Chapter 3: Premises and Equipment PREMISES General 3.2 Premises should be carefully maintained, ensuring that repair and maintenance operations do not present any hazard to the quality of products. They should be cleaned and, where applicable, disinfected according to detailed written procedures. |
Part 1 Chapter 3: Premises and Equipment PREMISES Production Area 3.10 Pipework, light fittings, ventilation points and other services should be designed and sited to avoid the creation of recesses which are difficult to clean. As far as possible, for maintenance purposes, they should be accessible from outside the manufacturing areas. |
Chapter 3: Premises and Equipment PREMISES Ancillary Areas 3.32 Maintenance workshops should as far as possible be separated from production areas. Whenever parts and tools are stored in the production area, they should be kept in rooms or lockers reserved for that use. |
Chapter 3: Premises and Equipment PREMISES EQUIPMENT 3.35. Repair and maintenance operations should not present any hazard to the quality of the products. |
4 Premises 4.9 (略) Floor drains in lower grade cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be regularly cleaned, disinfected and maintained. |
Manufacture of Herbal Medicinal Products Premises & Equipment Storage areas 3. Special attention should be paid to the cleanliness and maintenance of the storage areas particularly when dust is generated. |
12. Premises General 12.6 Premises should be carefully maintained, and it should be ensured that repair and maintenance operations do not present any hazard to the quality of products. |
Ancillary areas 12.13 Maintenance workshops should if possible be separated from production areas. Whenever parts and tools are stored in the production area, they should be kept in rooms or lockers reserved for that use. |
Production areas 12.28 Pipework, light fittings, ventilation points and other services should be designed and sited to avoid the creation of recesses that are difficult to clean. As far as possible, for maintenance purposes, they should be accessible from outside the manufacturing areas. |
4. Premises 4.9 (略) Floor drains in lower-grade cleanrooms should be fitted with traps or water seals designed to prevent backflow and should be regularly cleaned, disinfected and maintained. |
| 概 要 | 対象医薬品:全般 建物について以下の要件があります。 ・実施される作業にふさいわしいように保守管理すること。 ・建物の配置及び設計は、交叉汚染、じん埃又は汚れの蓄積及び( 一般的に)製品品質への悪影響を回避するために有効な洗浄や保守管理を可能とすること。 |
対象医薬品:全般 建物について以下の要件があります。 ・補修及び保守管理作業が製品の品質にいかなる危険も示さないことを保証するよう、建物は注意深く維持管理されること。 ・詳細な文書化された手順に従い清掃し、該当する場合には消毒すること。 |
対象医薬品:全般 製造区域について以下の要件があります。 ・配管、照明取り付け具、換気及び他のサービス供給箇所は、清掃しにくい窪みの形成を回避するように設計及び配置すること。 ・保守の目的の為、できる限り製造区域外から到達可能であること。 |
対象医薬品:全般 付随区域について以下の要件があります。 ・保守管理の作業場は、製造区域からできるだけ離されていること。 ・部品及び工具が製造区域で保管される場合には常に、それらはその用途に専用の部屋又はロッカーの中で保管すること。 |
対象医薬品:全般 設備について以下の要件があります。 ・修復及び保守管理作業は製品品質に対し、いかなる危険も示さないこと。 |
対象医薬品:無菌医薬品 以下の要件があります。 ・トラップ又はウォータシールについて、保守管理すること。 |
対象医薬品:植物性医薬品 以下の要件があります。 ・保管区域の清浄性及び的確な保守管理には特別な注意を払うこと。特に埃が発生する場合は、注意すること。 |
対象医薬品:全般 以下の要件があります。 ・製造工程、包装工程、保管工程で使用される全ての建物は好ましい状態になるように保全されること。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 原則と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 全般事項 3.2と同じ内容となります |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 製造区域 3.10と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.32と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 設備 3.35と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 保管区域 3と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 原則と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 全般事項 3.2と同じ内容となります |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 製造区域 3.10と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 建物 付随区域 3.32と同じ内容となります。 |
対象医薬品:全般 別紙(1) PIC/S GMP ガイドライン パート1 第3章 建物及び設備 設備 3.35と同じ内容となります。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |
対象医薬品:植物性医薬品 別紙(6) PIC/S GMP ガイドライン アネックス7 植物性薬品の製造 建物 保管区域 3と同じ内容となります。 |
対象医薬品:全般 以下の要件があります。 ・建物は注意を払って維持すること。 ・修理及び保守作業は、製品の品質に危害を及ぼさないこと。 |
対象医薬品:全般 以下の要件があります。 ・メンテナンスワークショップ(保守エリア)は、製造エリアとはできるだけ分離しておくこと。 |
対象医薬品:全般 以下の要件があります。 ・配管、照明器具、換気口その他の設備は、可能な限り製造エリア外からのアクセスにより保守を行うようにすること。 |
対象医薬品:無菌医薬品 別紙(2) PIC/S GMP ガイドライン アネックス1 無菌医薬品の製造 4 建物 4.9と同じ内容となります。 |